
Chapter 1 Introduction To Medicinal Chemistry
Medicinal chemistry is an interdisciplinary field of study which combines aspects of organic chemistry, physical chemistry, pharmacology, microbiology, biochemistry, as well as computational chemistry including bioinformatics and chemo informatics.
Medicinal chemistry is defined as an interdependent science that is a combination of applied (medicine) and basic (chemistry) sciences. It encompasses the discovery, development, identification, and interpretation of the mode of action of biologically active compounds at the molecular level.
It can be viewed as the melting pot of synthetic chemistry and molecular pharmacology that emphasizes the study of SAR of drug molecules; it therefore requires a clear understanding of both chemical and pharmacological principles. Drugs may be from natural origin or obtained through organic synthesis.
They are chemicals used for medicinal purposes. They interact with complex chemical systems (targets) of humans or animals. Medicinal chemistry is concerned with this interaction, focusing on the organic and biochemical reaction of drug substances with their targets.
The art of medicinal chemistry has enriched greatly from developments in the areas of organic chemistry, biology, biophysical/biochemical methods, and computational tools. While opportunities are enormous, advancing a drug candidate from bench top to clinic is associated with challenges as well, and a good understanding on both these aspects would significantly accelerate drug discovery process.
A drug molecule thus synthesized and clinically tested needs to be “developed” in suitable formulation before it comes to the shelves of the chemists for patient’s consumption purpose.
Read and Learn More Medicinal Chemistry III Notes
Medicinal chemistry continues to play a major role in drug research and development, taking advantage of newer techniques and increased knowledge of different branches of related sciences.
Important Techniques Used in Drug Development and Medicinal Chemistry:
- Quantitative structure activity relationship (QSAR)
- Molecular modeling
- Virtual screening and docking
- Drug target binding forces
- Combinatorial library design Bioinformatics
- Structure based drug design
- Peptidomimetics
- Analog design
- Rational drug design
- Chirality
- Study of structural basis for toxicity
- Use of natural products as drug leads
History And Development Of Medicinal Chemistry
Roots of Medicinal chemistry can be traced back to ancient folk medicine and early natural product chemistry. Since it serves as the link between chemical structure and observed biological activity, medicinal chemistry emerged about 100 years ago as a special discipline aiming to explore such relationships via chemical modification and structural mimicry of nature’s materials, particularly with the aim of enhancing the efficacy of substances thought to be of therapeutic value. Just as in all fields of science, the history of medicinal chemistry is comprised of the ideas, knowledge, and available tools that have advanced contemporary knowledge.
The Nineteenth Century Age of Innovation and Chemistry:
There is a long history of plants being used to treat various diseases. They figure in the records of early civilizations in Babylon, Egypt, India and China. The therapeutic properties of plants were described by the ancient Greeks and by the Romans and are recorded in the writings of Hippocrates, Dioscorides, Pliny and Galenus. Some metals and metal salts were also used at this time. In the middle ages various Materia Medica and pharmacopoeias brought together traditional uses of plants.
Exploration in the seventeenth and eighteenth centuries led to the addition of a number of useful tropical plants to those of European origin. Improved communications, between practitioners in eighteenth and nineteenth centuries resulted in progressive removal of preparations that were either ineffective or too toxic from herbals. It also led to a more rational development of new drugs.
The nineteenth century saw the beginnings of modern organic chemistry and consequently of medicinal chemistry. Their development is intertwined. Chemists throughout Europe refined and extended the techniques of chemical analysis. The synthesis of acetic acid by Kolbe in 1845 and of methane by Berthelot in 1856 set the stage for organic chemistry.
The emphasis was shifted from finding new medicaments from the vast world of plants to the finding of active ingredients that accounted for their pharmacologic properties. The isolation of a number of alkaloids including morphine, quinine and atropine from crude medicinal plant extracts was part of the analytical effort to standardize drug preparations and overcome fraud.
General anesthetics were introduced in surgery from 1842 onwards (diethyl ether, nitrous oxide and chloroform). Antiseptics such as iodine (1839) and phenol (1860) also made an important contribution to the success of surgery.
Many of the developments after the 1860s arose from the synthesis of compounds specifically for their medicinal action. Although the use of willow bark as a pain-killer was known to the herbalists, the analgesic activity of its constituent, salicin and of salicylic acid was developed in the 1860s and 1870s.
Once ideas of chemical structure were formulated in the mid-nineteenth century, the first theories of the relationships between chemical structure and biological activity began to emerge. Thus Crum-Brown and Fraser (1869) noted that a ‘relationship exists between the physiological action of a substance and its chemical composition! leading to the idea that cells can respond to the signals from specific molecules.
On the basis of observations that certain dyes selectively stained micro-organisms, Ehrlich in the 1890s put forward the idea that there were specific receptors for biologically active compounds – ‘lock and key’ relationships. The work of Meyer and Overton (1899-1901) to relate a physical property (the oil water distribution coefficient) to biological activity (anaesthesia) were the first rudimentary QSAR.
Another quantitative measurement that was made was the chemotherapeutic index, which was the ratio of the minimum curative dose to the maximum tolerated dose (CD50/LD50).
The Twentieth Century Age of Innovation and Chemistry:
Several different aspects of medicinal chemistry were developed in parallel through the second half of the twentieth century. Diseases of protozoal and spirochetal origin responded to synthetic chemotherapeutic agents. Interest in synthetic chemicals that could inhibit the rapid reproduction of pathogenic bacteria and enable the host organism to cope with invasive bacteria was dramatically increased.
The observation by Woods and Fildes in 1940 that the bacteriostatic action of sulfonamide-like drugs was antagonized by p-aminobenzoic acid, was one of the early examples in which a balance of stimulatory and inhibitory properties depended on the structural analogies of chemicals.
Together with the discovery of penicillin by Heming in 1929 and its subsequent examination by Florey and Chain in 1941 led to a water soluble powder of much higher antibacterial potency and lower toxicity than those of previously known synthetic chemotherapeutic agents. With the discovery of a variety of highly potent anti-infective agents, a significant change was introduced into medical practice.
Developments Leading to Various Medicinal Classes:
Psychiatrists have been using agents that are active in the central nervous system for hundreds of years. Stimulants and depressants were used to modify the mood and mental states of psychiatric patients. Amphetamine, sedatives, and hypnotics were used to stimulate or depress the mental states of patients. The synthesis of chlorpromazine by Charpentier ultimately caused a revolution in the treatment of schizophrenia.
The first pure hormone to be isolated from an endocrine gland was epinephrine, which led to further molecular modifications in the area of sympathomimetic amines. Subsequently, norepinephrine was identified from sympathetic nerves. The development of chroma- tographic techniques allowed the isolation and characterization of a multitude of hormones from a single gland. Various techniques emerged subsequently to take process of drug development to new heights.
The study of medicinal chemistry has certain defined objectives as mentioned below:
- Physico-chemical properties of drug molecules in relation to drug ADME.
- Chemical basis of pharmacology and therapeutics.
- Fundamental pharmacophores for drugs used to treat disease.
- Structure activity relationship (SAR) in relation to drug-target interactions.
- Chemical pathways of the drug metabolism.
- Application to making drug therapy decisions.
The Medicinal Chemist would strive hard to convert a chemical into suitable lead molecule and then into a drug using various tools and approaches of medicinal chemistry.
Physicochemical Properties In Relation To Biological Action
Classification of drugs:
Drugs are chemical substances which can generally be classified based on their therapeutic use as follows:
- Drugs which act upon various physiological functions of the body (Pharmacodynamic agents) e.g. sedatives, analgesics, antipyretic and antirheumatic agents, antipsychotic, antihistaminic and anti-allergic drugs, anti-inflammatory agents, diuretics, cardiovascular agents and drugs acting on the heart, adrenergic and cholinergic agents and drugs acting on the gastrointestinal tract (GIT), etc.
- Drugs acting on Central Nervous System (CNS agents) e.g. antidepressant drugs, anaesthetic, anticonvulsants, antipsychotics, hypnotics and sedatives etc.
- Drugs that are used to fight pathogenic organisms (chemotherapeutic agents): e.g. sulfonamides, antibiotics, anti-infective agents, antimicrobial, anti-amoebic, antifungal agents, antiviral agents, anticancer agents, anti-malarial agents, etc.
- Supplement agents: e.g. Vitamins, dietary supplements, etc.
It is an established fact that “structure” is the fundamental aspect of drug molecule which affects both its pharmacodynamic and pharmacokinetic properties. In the given Unit, we will discuss about how structural features of the drugs impact its pharmacological actions.
Whenever a drug molecule enters the body, it interacts with one or more biomolecules found in the extracellular fluid, in the cell membrane, and within cells. The type and the extent of this interaction will depend on the kind and number of chemically reactive functional groups and the polarity of the drug molecule. The drug-protein interaction does not involve covalent bonds that are relatively stable at body temperatures.
Instead, weak forces such as ionic bonds, hydrogen bonds, Van der Waals forces, dipole-ion, and dipole- dipole forces are involved. A biological response is produced by the interaction of a drug with a functional or an organized group of molecules, generally referred as receptor site (chemoreceptive substance).
The physical and chemical properties (physicochemical properties) which affect biological action of drug are as follows:
- Ionization
- Partition coefficient (Log P)
- Solubility
- Protein binding
- Chelation
- Bioisosterism
- Hydrogen bonding
- Optical and Geometrical Isomerism
Ionization
Ionization is the process of formation of ions, in which an atom or a molecule possesses a negative or positive charge by gaining or losing electrons, often in conjunction with other chemical changes.
The first three stages of the pharmacokinetic process [absorption, distribution, metabolism and excretion (ADME)] are:
- Liberation: Release of the medicinal drug from a formulated medicine.
- Absorption: Movement of the liberated drug into the bloodstream.
- Distribution: Passage of the drug from the bloodstream to body tissues and organs.
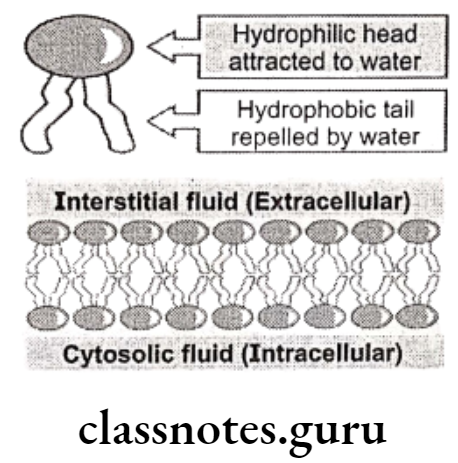
Molecular compounds are less soluble in water than in non-polar solvents, while ionic compounds are more soluble in water than in non-polar solvents. In the body there are two distinct environments. One, such as blood plasma, is aqueous and so ionic compounds are more likely to be soluble in blood than are molecular compounds. The other is cell membrane which is mainly composed of non-polar lipids (phospholipids, glycolipids and cholesterol).
The central part of the membrane, therefore, consists mainly of long chain hydrocarbons and so molecular compounds are more likely to be soluble in cell membrane than are ionic compounds. An effective drug needs to be sufficiently soluble in water to dissolve in blood plasma and be carried around the body while also being sufficiently soluble in non-polar lipids to pass through cell membranes into cells.
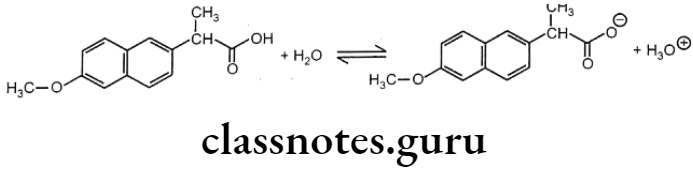
Most drug molecules ionize in aqueous solution to give weakly acidic or basic solutions. Naproxen is a non-steroidal anti-inflammatory drug (NSAID). It is used to relieve pain and inflammation in diseases like rheumatic disease, sprains, strains, backache, gout, and menstrual pain.
A naproxen molecule has a carboxylic group and so it is a weak acid. It ionizes in water to give an equilibrium solution that contains a mixture of unionized molecules, carboxylate ions and hydroxonium ions.
Aspirin, fenoprofen and penicillin-G (benzylpenicillin) are the drug molecules that also contain carboxylic acid groups. All three compounds partially ionize in aqueous solution.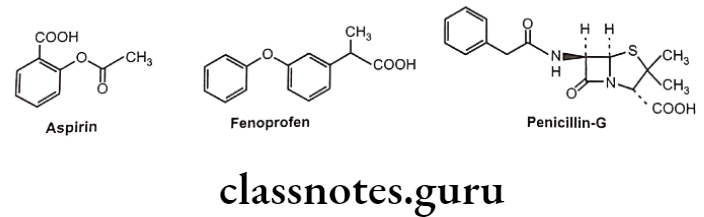
Diphenhydramine is a first generation antihistamine used to treat allergic symptoms though it does have as sedating effect. More recently developed antihistamines are not sedating. The sedating properties of diphenhydramine explain its use to treat insomnia and travel sickness. Diphenhydramine molecule has an amine group and hence it is a weak base. It ionizes in water to give an equilibrium solution that contains a mixture of unionized molecules, quaternary ammonium ions and hydroxide ions.
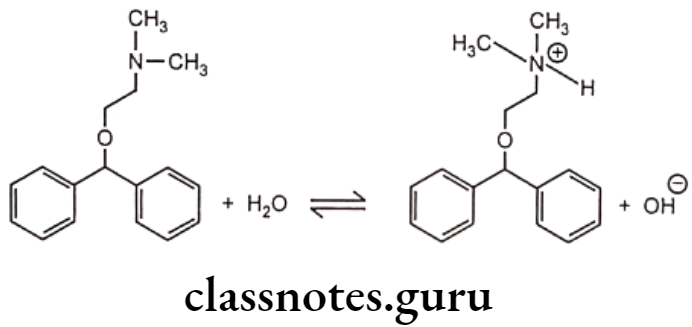
Other drugs which are bases include adrenaline, ephedrine, atropine and tetracycline. The rate of absorption depends on concentration of unionized form. Accumulation of an ionized drug in the body compartment is called as ion trapping. Drug ionization depends on pH and its pKa.
Most drugs can be classified as acids or bases as a large number of drugs can behave as either acids or bases, as they circulate in the patient in different dosage forms and end up in systemic circulation. Acid-base properties of a drug can considerably influence its bio-distribution and partitioning characteristics.
For the definition of acids and bases, the model used in pharmacy and biochemistry was developed independently by Lowry and Bronsted. An acid is a proton donor and a base is a proton acceptor.
Percent Ionization
Percent ionization is defined as the amount of a weak acid that exists as an ions at a particular concentration. Ionized drugs are water soluble and unionized drugs are lipid soluble.
HA Unionized acid, H2O= base, H,O= conjugate acid, A = ionized conjugate base.
B = Unionized base, H2O= acid, BH = conjugate acid, OH Conjugate base Change in pH changes the degree of ionization.
Examples:
- In Phenytoin, pKa = 8.3 i.e. at pH 8.3, it is 50% ionized. To ionize completely, its pH is raised to 12 by NaOH and complete ionization is ensured by maximum H2O solubility.
- In Aspirin, pKa= 3.5, pH = 1, it is 99% more unionized. It is lipid soluble and easily absorbed through stomach.
Acid-conjugate base:
Representative examples of pharmaceutically important acidic drugs are listed. Each acid, or proton donor, yields a conjugate base. Conjugate bases range from the chloride ion [reaction (a)], which does not accept a proton in aqueous media, to ephedrine [reaction (h)], which is an excellent proton acceptor.
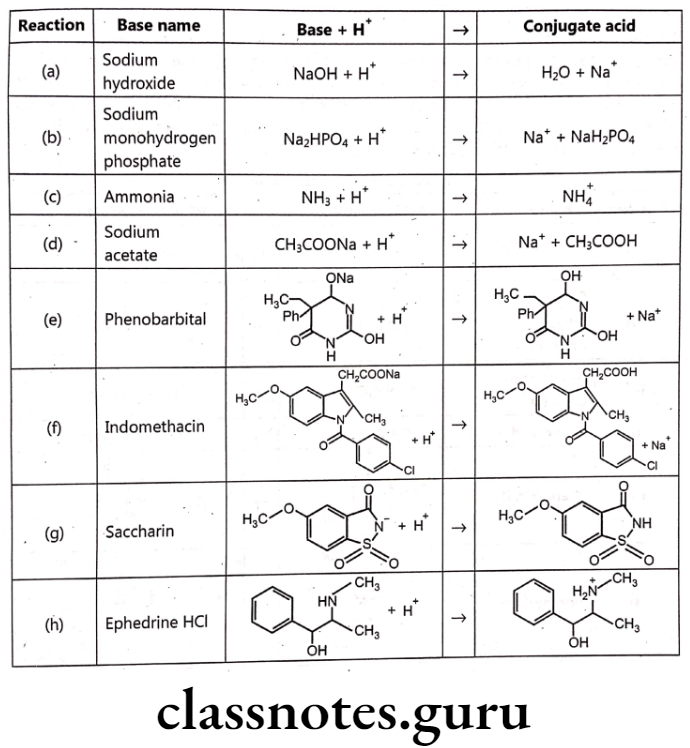
Base-conjugate acid:
The Bronsted-Lowry theory defines a base as a molecule that accepts a proton. The product resulting from the addition of a proton to the base is the conjugate acid. Pharmaceutically important bases are listed. There are a variety of structures, including the easily recognizable base sodium hydroxide [reaction (a)]; the basic component of an important physiological buffer, sodium monohydrogen phosphate [reaction (b)], which is also the conjugate base of dihydrogen phosphate; ammonia [reaction (c)], which is also the conjugate base of the ammonium cation [reaction (c)]; sodium acetate [reaction (d)], which is also the conjugate base of acetic acid [reaction (d)] in the enolate form of phenobarbital [reaction (e)], which is also the conjugate base of phenobarbital [reaction (e)]; the carboxylate form of indomethacine [reaction (f)], which is also the conjugate base of indomethacine [reaction (f)]; the imidate form of saccharin [reaction (g)], which is also the conjugate base of saccharin [reaction (g)] in and the amine ephedrine [reaction (h)], which is the conjugate base of ephedrine HCI. 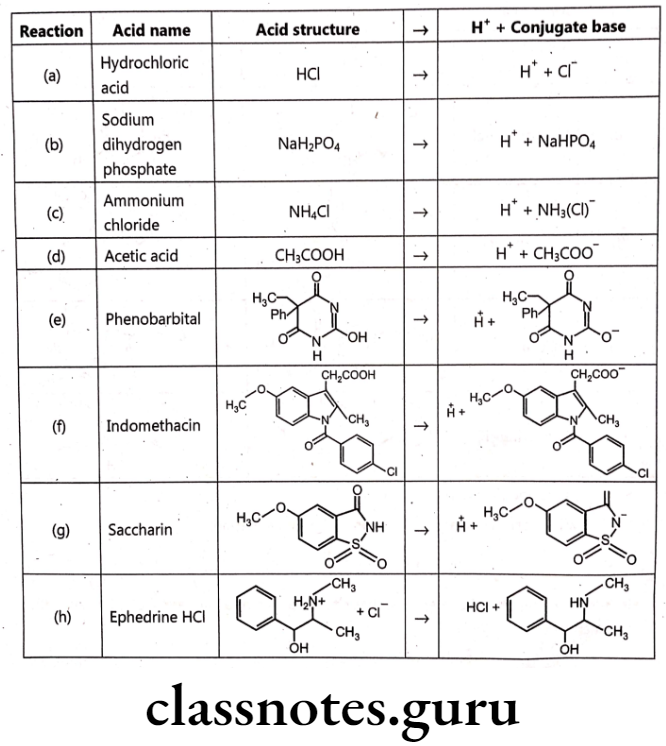
Since, pKa = -log Ka and pH = -log [H3O]*.
The pH will be calculated according to the following equation:
It is now common to express the basicity of a chemical in terms of pka using the following equation:
Examples of calculations requiring the pKa (a):
To determine the ratio of ephedrine to ephedrine HCI, (pKa 9.6) in the intestinal tract at pH 8.07. Using Eq. (3) we get,
The number 1.6 whose log is 0.025, meaning that there are 25 parts ephedrine for every 1000 parts ephedrine HCI in the intestinal tract whose environment is pH 8.0.
To determine the pH of a buffer containing 0.1 mol/lit CH3COOH (pKa 4.8) and 0.08 mol/lit CH3COONa. Using Eq. (3) we get,

Importance of ionization of drugs:
- Unionized form of a drug can partition across biological membranes as the unionized form is lipophilic.
- Ionized form tends to be more water soluble as it is important for drug administration and distribution in plasma.
- Lower the pH with respect to pKa, greater will be the fraction of protonated drug which may be charged or uncharged.
- Weak acids are more lipid-soluble, because they are uncharged and hence, this form passes through biological membranes more rapidly. Acidic pH provides a proton to weak acids to form uncharged species.
RCOO + H* → RCOOH - Similarly alkaline pH will cause weak bases to lose a proton to form uncharged species.
Partition Coefficient
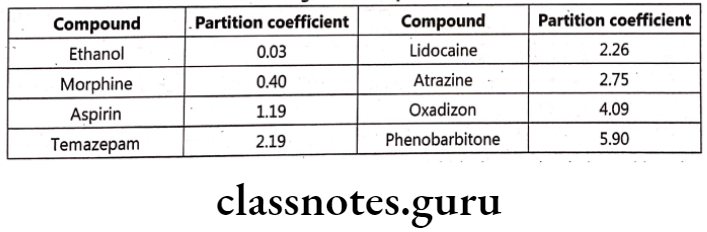
Partition coefficient (P) finds importance as it directly influences drug transport and drug distribution. It is defined as ratio of concentrations of compounds in two phases of a mixture of two immiscible liquids at equilibrium. E.g. Phenobarbitone has a high lipid / water partition coefficient of 5.9, whereas Thiopentone sodium has a chloroform / water partition coefficient of about 100 which states that the later is highly soluble in lipid. It is defined as equilibrium constant of drug concentration in two phases.
It is difficult to measure partition coefficient in a living system. It is usually estimated in vitro by using octanol as a lipid phase and phosphate buffer (pH 7.4) as the aqueous phase. Partition coefficient is dimensionless and its logarithm (log P) is widely used as the measure of lipophilicity. Factors affecting partition coefficient are:
- pH
- Co-solvents
- Surfactant
- Complexation
Log P can be determined by shake flask method in which drug molecule is partitioned between known quantity of octanol and that of buffer or water. The concentration of drug is determined by UV spectroscopy method. This method, however, is a cumbersome method. Another method includes HPLC method, in which compounds with known log P are injected onto a C18 reverse phase HPLC column followed by unknown compounds.
Applications of Partition Coefficient
- Solubility of drugs in water and other solvents and in mixture of solvents can be predicted.
- Drug absorption in vivo can be predicted.
- SAR for a series of drugs can be studied.
- Log P is an indicator of lipophilic and hydrophilic character of a drug molecule.
Solubility
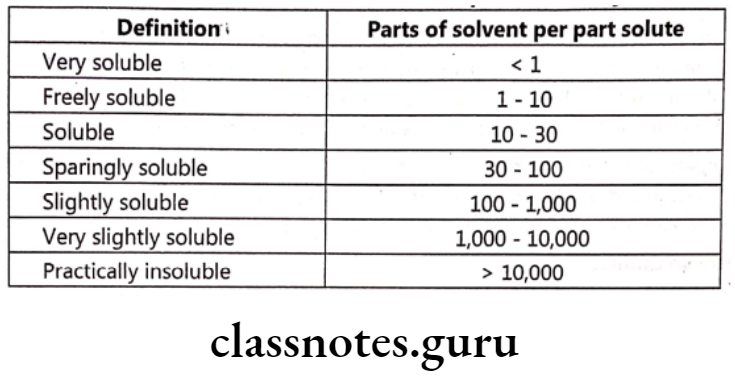
Solubility is defined as the ability of a substance (solute) to enter into a bulk medium (solvent) and form a homogenous solution. Different compounds have different solubilities depending on various factors. Solubility refers to the concentration of dissolved solute, which is in equilibrium with solid solute at a given temperature. Sufficient solubility and good membrane permeability are prime factors affecting oral absorption.
It has been said that low solubility is at the top of the list of undesirable properties of a potential medicinal drug. So measurement of solubility and, if necessary, modification of a compound to alter its solubility without affecting its therapeutic properties is important.
Equilibrium solubility:
The equilibrium solubility of a compound is the concentration of a saturated solution in contact with an excess of undissolved solid.
Traditionally, excess compound is shaken with a solvent until a saturated solution is produced with an excess of the solid compound present. It is an accurate but very time-consuming method.
Kinetic solubility:
The kinetic solubility in water of a drug that is a weak acid is determined by:
- dissolving the acid in a known volume of base, e.g. potassium hydroxide solution;
- adding an acid, e.g. hydrochloric acid, until a precipitate is detected (the UV sensor ‘sees’ the first formation of cloudiness).
Rather than solubility, logs is usually calculated.
Aqueous solubility depends upon the following facts:
- Buffer and Ionic strength
- Polymorphism and purity of the sample
- PH
- Super saturation
- Thermodynamic vs. kinetic solubility
Aqueous solubility decreases with corresponding increase in various physicochemical properties like boiling point, viscosity, surface activity and partition coefficient across a homologous series. Solubility of a drug can be increased or decreased by derivatization e.g. water soluble methyl predinisolone acetate is transformed to methyl predinisolone sodium succinate which is water soluble.
Similarly, slightly soluble chloramphenicol can be converted into insoluble chloramphenicol palmitate. Solubility not only depends on the solute and solvent, but also to a larger extent on temperature, pressure and pH. Solubility of a molecule can also be expressed in terms of its affinity or philicity and repulsion or phobicity for either an aqueous (hydro) or lipid (lipo) solvent.
Various types of bonds like London forces, hydrogen bonds, dipole-dipole, etc. are responsible for keeping atoms and molecules together which are responsible for maintaining solubility. Following intermolecular attractive forces are responsible for solubility:
- Vander Waals’ forces: These are weakest (0.5 -1.2 kcal/mole) of all the forces resulting from induced dipole and occur between non-polar groups e.g. hydrocarbons.
- Dipole-dipole interaction: These are strong (1.2 to 10.8 kcal/mole) forces arising ionically between dipoles created due to an electronegative atom attached to carbon atom. E.g. Hydrogen bond that contributes for hydrophilicity.
- Ionic bonding: These arise mostly in inorganic compounds and their salts and are comparatively strong (5 kcal/mole) bonds. The degree of ionization for any molecule is an important factor affecting solubility.
- Ion-dipole bonding: These are relatively strong bonds (1.2 to 5.2 kcal/mole) similar to ionic interactions and arise electrostatically between a cation/anion and a dipole. These forces depend on distance and temperature.
- Covalent bond: Molecular bond that involves the sharing of electron pairs between atoms is a covalent bond and is greatly affected by electronegativity of atoms which determines the chemical polarity of the bond. These are the strongest of all the bonds and may vary from 50 kcal/mol to 110 kcal/mol depending on the elements involved.
Many methods have been tried and tested to improve solubility of drug molecules which include:
- Use of co-solvents (like glycols, sorbitol, ethanol etc.) for highly lipophillic molecules.
- Use of surfactants (like Tween 80) which being amphiphillic (polar head and non- polar tail), enhances solubility by forming micelles which partitions the non-polar drug into its hydrophobic core.
- Use of hydrotropes, which are organic molecules and when added in water, forms loose aggregates. The drug gets dissolved in the matrix of the hydrotrope.
- Structural alternations of drug molecule (e.g. Carrier linked prodrugs)
- Use of complexing agents which uses various forces like London dispersion forces, hydrogen bonding and hydrophobic interactions to form a complex with drug molecule leading to enhanced solubility. e.g. Cyclodextrin complexes.
- Changing the pH of aqueous solution of drug molecule so that ionization pattern is affected. Solubility increases with ionization as ions are more soluble in aqueous media than neutral molecules. Hence, pH of the solution can be increased or decreased depending on the nature of drug.
Factors Affecting Solubility:
- Temperature: Increase in temperature increases kinetic energy of the system, thereby increasing solubility. Heat energy gets converted to kinetic energy which causes the molecules of both solute and solvent to vibrate causing more interaction between the molecules.
- Pressure: Pressure doesn’t affect the solubility of solids and liquids to a great extent. However, the solubility of gases increases with increase in pressure.
- Particle size of solute: Smaller the particle size, more is the solubility. Small particle size results in large surface area and large surface area means more interaction with the solvent and hence, more dissolution. But extremely small particle sizes can lead to aggregation of the particles to form clumps which again decreases solubility.
- Polymorphism: Different forms of a compound have different solubilities. Generally, amorphous forms are more water soluble than crystalline materials as more surface area is available for solvent interactions due to finer particle size. Furthermore, in crystalline compounds, anhydrous forms are more soluble than hydrate forms as hydrate forms already contain bound water within their crystal lattice. Also, metastable forms of crystals have greater solubility than stable forms.
- Polarity: The polarity of solute as well as solvent can affect solubility of the solute. If a solute is polar in nature, it will dissolve only in a polar solvent. This is because of dipole-dipole interactions between the solute and the solvent molecules. The positive end of a solute molecule will have affinity for the negative ends of the solvent molecules and such interactions form a solution. Similarly, non-polar solutes will be soluble in non-polar solvents due to the presence of London dispersion forces. These are attractive forces that form temporary dipoles of atoms and molecules thus aiding solubility.
Some other factors include the nature of solute, nature of solvent, molecular size of solute, melting point of solid solutes, etc., which affect the extent of solubility of a compound in variable degrees.
Importance of Solubility:
- Solubility is of great concern as it is necessary that the drug must be in the solution form so that it can be absorbed by the body or have any biological activity.
- Drugs (mainly organic) with limited aqueous solubility won’t dissolve in the body fluids which decreases bioavailability.
- Not only lipophilicity plays an important role to cross the lipid bilayer of plasma membrane, but the drug must also dissolve in the aqueous portion of the cell for optimum absorption.
- Drugs must dissolve so that it can interact freely with biological targets.
Protein Binding
Binding of drug to protein may enhance distribution of drugs, or inactivate the drug by not enabling a sufficient concentration of free drug to target at receptor site or retard the excretion of such drug molecule. Interaction of drugs with protein may cause displacement of body hormones or change the configuration of protein or inactivates the drug biologically by forming a drug-protein complex.
Drug binding to a protein or multiple proteins mainly depends on nature (acidic or basic) of drug. Albumin is the most common protein that is involved in binding of drugs and comprises of more than half of blood proteins. Albumin can interact with both, acidic as well as basic drugs in the plasma by Van der Waals’ dispersion forces, hydrophobic bonding, hydrogen bonding and ionic interaction.
Protein binding values (% fraction bound) are normally given in terms of percentage of total plasma concentration of a drug that is bound to all plasma proteins. Drugs bind to protein either through hydrophobic interactions (e.g. binding of Ca2+ to target protein) or through self-association in which drug may self dissociate to form dimers, trimers or aggregates of larger size.
Many times binding to plasma proteins is reversible, and the concentration of the free and bound drug at equilibrium may be expressed as:
Free drug + Free protein = Drug-protein complex
Pharmacokinetic importance of protein binding:
- Drug-protein binding influences the distribution equilibrium of the drug.
- Plasma proteins exert a buffer and transport function in the distribution process.
- Only free and unbound drug acts can leave the circulatory system and diffuse into the tissue.
- Protein binding is affected by the presence of diseases. e.g. drugs like dapsone, morphine show decreased extent of protein binding in liver diseases, whereas drugs like barbiturates and sulphonamides show less binding in renal diseases.
Protein binding can be determined by many methods like dialysis, ultracentrifugation, ultra filtration, electrophoresis etc.
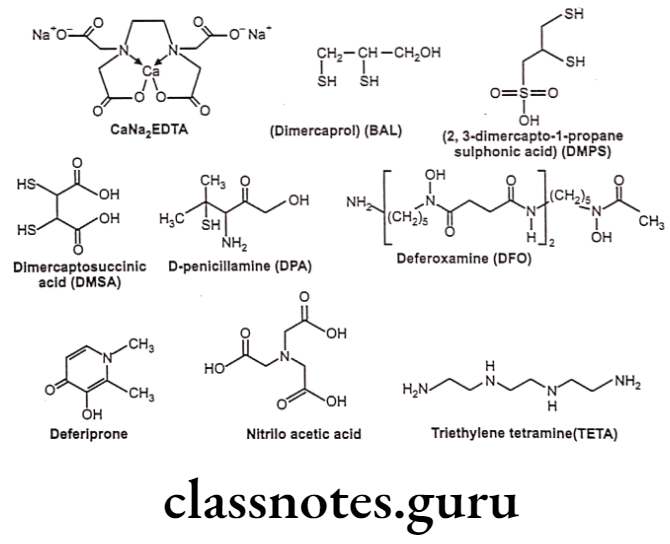
Chelation
Chelating agents are organic or inorganic compounds capable of binding metal ions to form complex ring-like structure called ‘chelates’. Chelating agents possess “ligand binding atoms that form either two covalent linkages or one covalent and one co-ordinate or two co-ordinate linkages in the case of bidentate chelates.
Mainly atoms like S, N and O function as ligand atoms in the form of chemical groups like -SH, -S-S, -NH2, NH, -OH, -OPO3H, or >C=O. Bidentate or multidentate ligands form ring structures that include the metal ion and the two-ligand atoms attached to the metal. Chelation is a result of a donor-acceptor mechanism (electrons or electron pair) or Lewis acid-base reaction (protons) leading to formation of complexes or coordination compounds.
Any non-metallic atom or ion (in free form or in neutral molecule or in ionic compound) that can donate an electron pair may serve as the donor. Any metallic ion or a neutral molecule may accept a pair of electrons and acts as acceptor. In addition, intramolecular forces can also be involved in the formation of complexes. Complexes may be divided broadly into two classes depending on whether the acceptor compound is a metal ion or an organic molecule.
Many a times, an ideal chelator in vitro might not prove so in vivo, either due to the toxicity considerations or due to the presence of endogenous substances (haemoglobin, cytochromes, etc.) that can also chelate metal ions and thus leads to competition. Also, pH appears to be an important factor influencing complex formation and stability.
Most chelating agents are unstable at low pH, whereas at high pH metals tend to form insoluble hydroxides which are less accessible to chelating agents. This feature becomes significant in pathological conditions leading to acidosis or alkalosis. Optimally effective chelation can be achieved by virtue of some combination of the basic properties of both the metal ions, chelating agents and the resulting metal complex.
Applications of chelation:
- It is significantly involved in biological system and to some extent in explaining drug action.
- Can lead to effective antidotes for organic arsenicals, treatment of poisoning due to antimony, gold and mercury, etc. e.g. Dimercaprol.
- Penicillamine is an effective antidote for treatment of copper poisoning because it forms water-soluble chelate with copper and other metal ions.
- 8-Hydroxyquinoline and its analogue acts as antibacterial and antifungal agent by complexing with iron and copper.
Bioisosterism
Longmuir coined the term isosterism. When two molecules or molecular fragments containing an identical number and arrangement of electron will have similar properties then they are termed as isosteres. Isosteres should be isoelectric i.e. they should possess same total charge. The phenomenon of isosterism is important because the biological characteristics of isosteres appear to be similar.
Bioisosterism is defined as the phenomenon by which compounds or groups that possess similar molecular shapes and volumes, approximately same electronic distribution and similar physical properties exhibits similar biological activities.
Classification:
Bioisosteres are classified into the following two types:
- Classical bioisosteres
- Non-classical bioisosteres
Classical bioisosteres:
- Classical bioisosteres have similarities in shape and electronic configuration of atoms, groups, and molecules, which they replace. They have similarities of shape and electronic configuration of atoms, groups and molecules which they replace.
- The classical bioisosteres may be,
Univalent atoms and groups:
Cl, Br, F, H, OH, NH, CH3 for H, SH and CF3
Bivalent atoms and groups:
Trivalent atoms and groups:
-CH=, N =, p =, – AS =
Tetravalent atoms and groups:
=N*, C=,=P*=,=As*=
Ring equivalents:
Non-classical Bioisosteres:
They do not follow the steric and electronic definition of classical isosteres. They do not have the same number of atoms as replacement. They have one of the following characteristic features, such as
- Electronic properties
- Physicochemical properties
- Spatial arrangements
- Functional moiety critical for biological activity
Exchangeable groups: Exchangeable group isosterism in which the properties of discrete functional elements are emulated. Eg. Carboxylic acid functionality can be exchanged with tetrazole or sulphonamido groups.
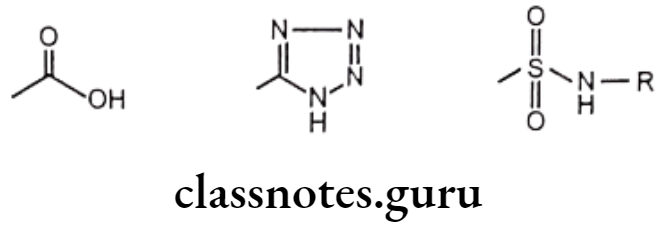
Cyclic versus non-cyclic structure:
Hydrogen Bonding
Among the secondary forces, hydrogen bonding is one of the most important forces that affects the physical property of the compound. Hydrogen bonds are created because of groups like -OH, -NH, etc. which can form intermolecular or intramolecular hydrogen bonds e.g. water, salicylic acid, o-nitro phenol etc. Hydrophobic bonds are seen between non-polar regions of the receptor and the drug e.g. isopropyl moiety, of a drug fits into a hydrophobic cleft on the receptor composed of hydrocarbon chains of amino acids valine, isoleucine and leucine.
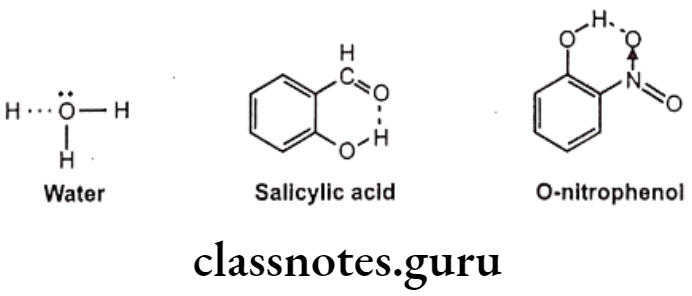
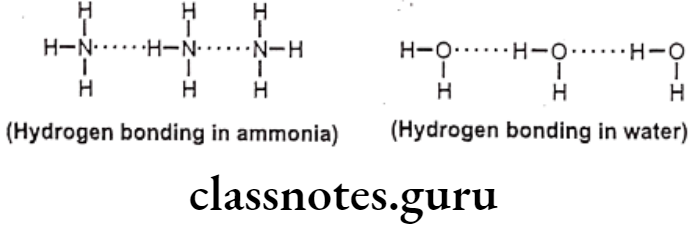
Hydrogen bonds are generally, of two types, one, intermolecular hydrogen bonding and other one is intramolecular hydrogen bonding. The intermolecular hydrogen bonding occurs between different molecules of same compound so as to form polymeric aggregate. This cause increases in boiling point of the compound as well as its water solubility.
The reason for increase in solubility is the formation of intermolecular hydrogen bonding with water e.g. ethanol shows higher boiling point and higher solubility in water than dimethyl ether even though both have the same molecular weight. And the intramolecular hydrogen bonding occurs between different atoms of same molecule.
Such kind of bonding may result into ring like systems thus causing decrease in boiling point and water solubility. The reason for decrease in solubility is the restriction of possibility of intermolecular hydrogen bonding of the o-substituted groups with water that prevents association of the molecules to form an aggregate.
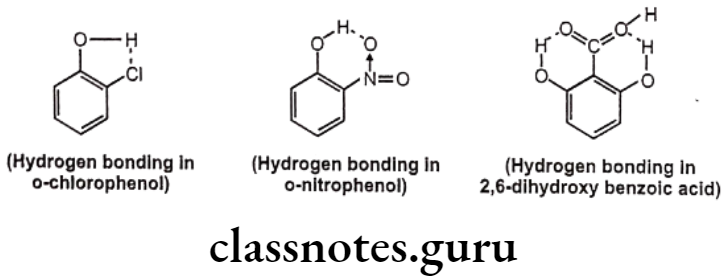
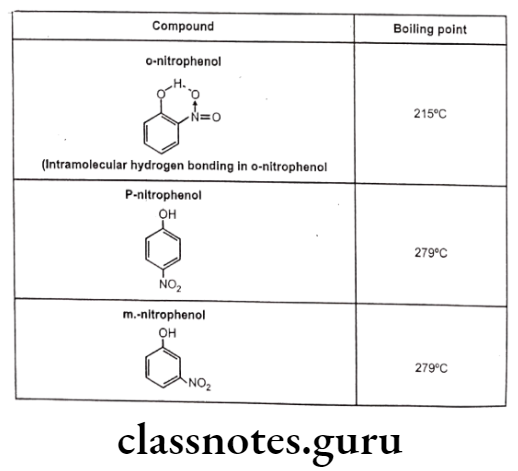
Effects of Hydrogen Bonding:
All physical properties such as boiling points, melting point, water solubility, etc. and several chemical properties like acid character, basic character, properties of carbonyl groups, etc. are affected by hydrogen bonding.
For example, Intermolecular hydrogen bonding cause association of several molecules of the same compound leading to increase in intermolecular forces and increased boiling point. The intramolecular hydrogen bonding cause chelation between the groups of same molecule thereby restricting the possibility of intermolecular hydrogen bonding and thus reduces melting point and boiling point.
Hydrogen bonding between the solvent and the solute increases aqueous solubility by many folds e.g. methanol and ethanol are highly soluble in water due to hydrogen bonding between molecules. Similarly, high solubility of polyhydric phenols and sugars may be due to availability of greater number of -OH groups for hydrogen bonding.
Compounds showing hydrogen bonding have higher surface tension and viscosity e.g. glycerol, glycol, sulphuric acid, sugar syrup, phosphoric acid, etc. Due to more number of -OH groups, the extent of hydrogen bonding is more in glycerol. So, it is more viscous than glycol. The tertiary structures of proteins and nucleic acids are due to hydrogen bonding.
In a-helices, hydrogen bonds are formed between hydrogen of polar N-H units with oxygen of polar C=O units. Also, the double strands of DNA are held together by hydrogen bonds. Replication of DNA depends on hydrogen bonds which selectively connect specific base pairs, as do the several steps by which the genetic message determines the specific order of amino acids in a protein.
Drug-Receptor interactions: Hydrogen bonding provides secondary binding force for drug-receptor interactions.
Isomerism
Isomers are the molecules of identical atomic compositions, but with different bonding arrangements of atoms or orientations of their atoms in space i.e., isomers are two or more different substances with the same molecular formula. Three types of isomerism are possible – Constitutional, Configurational, and Conformational. The terms configuration and conformation are often confused.
Configuration refers to the geometric relationship between a given set of atoms, for example, those that distinguish L- from D-amino acids. Interconversion of configurational alternatives requires breaking of covalent bonds. Conformation refers to the spatial relationship of every atom in a molecule. Interconversion between conformers occurs without covalent bond rupture, with retention of configuration, and typically via rotation about single bonds.
Geometrical Isomerism:
Stereochemistry, enantiomers, symmetry and chirality are impotent concepts in therapeutic and toxic effect of drug. The drug must possess a high degree of structural specificity or stereo selectivity. Many drugs show stereo selectivity because mostly the sites or targets are optically active biological macromolecules such as proteins, polynucleotides or glycolipids.
Geometrical isomers are stereoisomers having same molecular formula, same functional groups, same positions, but different orientation around a double bond or on a ring.
An important criteria to exhibit geometric isomerism is that the isomers cannot be interconverted through mere rotation around a single bond. The relative positions of atoms attached directly to multiple bonds are also fixed. For the double bond, cis- and trans- isomers result.
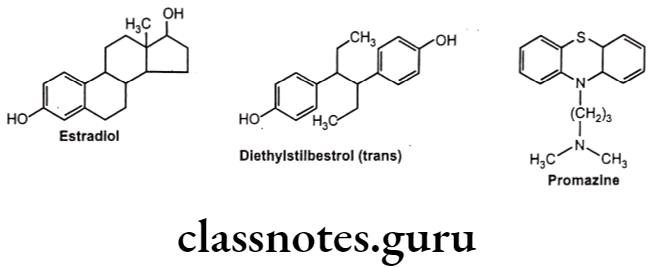
For example, diethylstilbestrol exists in two fixed stereoisomeric forms: trans- diethylstilbestrol is estrogenic, whereas the cis-isomer is only 7% active. In trans- diethylstilbestrol, resonance interactions and minimal steric interference tend to hold the two aromatic rings and connecting ethylene carbon atoms in the same plane.
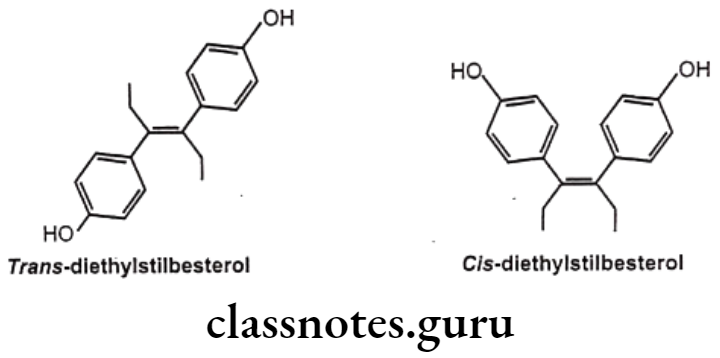
Geometric isomerism is represented by cis/trans isomerism resulting from restricted rotation due to carbon-carbon double bond or in rigid ring system. They are two or more coordination compounds which contain the same number and types of atoms, and bonds (i.e., the connectivity between atoms is the same), but which have different spatial arrangements of the atoms. Specific isomer gives specific response and hence different pharmacological responses are obtained for different isomers.
Optical Isomerism:
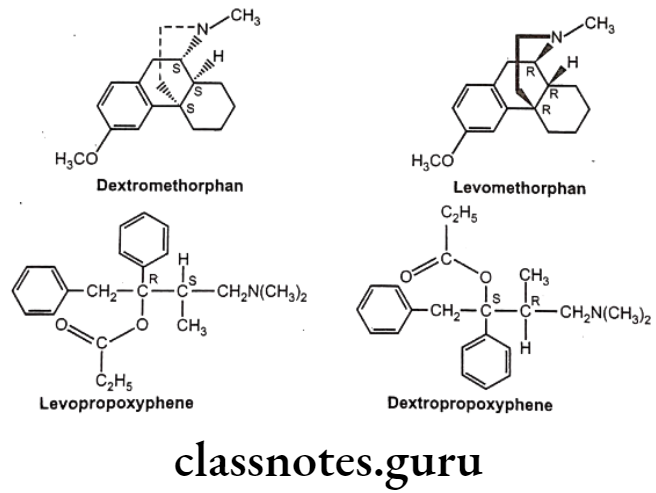
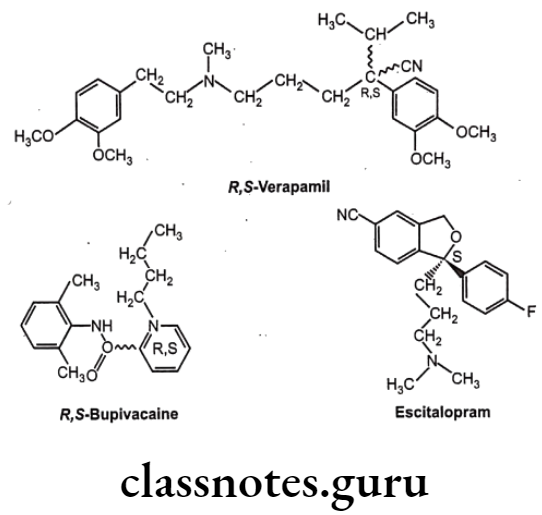
Optical isomers are non-superimposable mirror images of each other (called a pair of enantiomers) and differ in their optical activity. Many drugs come from natural sources like plants. They are usually chiral and are generally found only as single enantiomers in nature rather than as racemic mixtures. Example, synthetic drugs like Ibuprofen are chiral, one of its enantiomers has analgesic and anti-inflammatory properties, the other does not (it is pharmacologically inactive).
Stereoisomers differ in pharmacokinetic and pharmacodynamic properties. Pharmaco- kinetic differences resulting out of stereo-isomerism can be in absorption, e.g. L-Methotrexate is better absorbed than D-Methotrexate, Esomeprazole is more bioavailable than racemic omeprazole; in distribution, e.g. S-Warfarin is more extensively bound to albumin than R-Warfarin, hence it has lower volume of distribution.
Levocetrizine has smaller volume of distribution than its dextroisomer, D-Propranolol is more extensively bound to proteins than L-Propranolol; in metabolism like S-Warfarin is more potent and metabolized by ring oxidation while R-Warfarin is less potent and metabolized by side chain reduction, half life of S-Warfarin is 32 hours, while it is 54 hours for R-Warfarin.
Pharmacodynamic differences resulting out of stereoisomerism can be in pharmaco- logical activity and potency like L-Propranolol has ẞ-adrenoceptors blocking action while D- Propranolol is inactive; Carvedilol is a racemic mixture, the S(-) isomer is a nonselective ẞ- adrenoceptor blocker, while both S(-) and R(+) isomers have approximately equally α- blocking potency; S-Timolol is more potent a-blocker than R-timolol but both are equipotent ocular hypotensive agents.
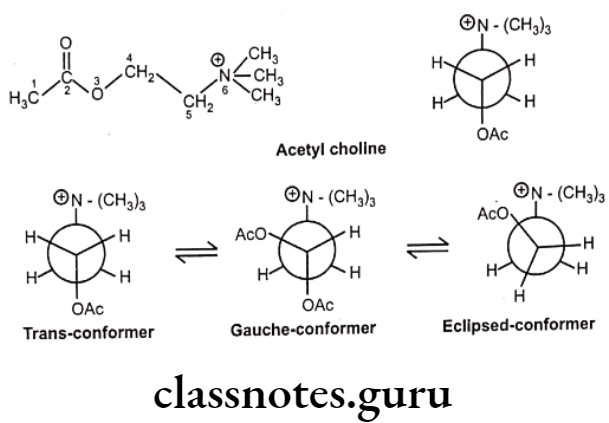
Conformational Isomerism:
Another form of isomerism is conformational isomerism which arises due to free rotation around carbon-carbon single bond. Conformational isomers (or conformers) are non- superimposable orientations. In order for a molecule to possess conformational isomers, it must possess at least one single bond that is not part of a ring system.
The reason for this restriction is that it is impossible to freely rotate a single bond within a ring system without breaking the ring in the process. The conformational flexibility of most open-chain neurohormones, such as acetylcholine, epinephrine, serotonin, histamine, and related physiologically active biomolecules, permits multiple biological effects to be produced by each molecule, due to their ability to interact in a different and unique conformation with different biological receptors.
For example, acetylcholine may interact with the muscarinic receptor of postganglionic parasympathetic nerves and with acetyl cholinesterase in the fully extended conformation and, in a different, more folded structure, with the nicotinic receptors at ganglia and at neuromuscular junctions.
In the structure of acetylcholine, the ethane bridge (i.e., atoms 4 and 5) between the ester oxygen and the quaternary nitrogen is a freely rotatable system and gives rise to a variety of different conformations. When the acetyl group and the quaternary nitrogen are situated 180° apart, the molecule is said to be in the trans or anti conformation.
Both a sawhorse representation and a Newman projection of the trans conformation are illustrated. Rotation of the trans Newman projection 60° in the counterclockwise direction gives rise to the gauche or skew conformation. Continued rotation by another 30° in the same direction provides the fully eclipsed conformation.
These rotations alter the spatial arrangement of the atoms without breaking a bond. Therefore, conformational isomers are not distinct molecules, but rather different orientations of the same molecule. Obviously, there are many more conformations possible.
Drug Metabolism
Metabolism (biotransformation) is a necessary phenomenon to eliminate drugs or other foreign compounds (xenobiotic) from body. During metabolism the lipophilic compounds are converted into hydrophilic one and are excreted out. Drug metabolism is also referred as detoxification process, (but prodrugs are exceptional to this) because of which toxic metabolites are wiped out of body.
Drug metabolism takes place in two phases:
- Phase 1 (Functionalization)
- Phase 2 (Conjugation)
Phase-1: Functionalization
Functionalization includes oxidation, reduction and hydrolysis. The purpose is to introduce a polar functional group like OH, COOH, NH2, SH, into xenobiotic molecules with the help of several enzymes.
This can be done in two ways:
- Direct introduction of functional group (aromatic /aliphatic hydroxylation)
- Modification or unmasking of existing functionalities.
e.g. Reduction of ketone and aldehyde to alcohol, oxidation of alcohols to acids, hydrolysis of esters and amide to acid, amines and OH groups, reduction of azo and nitro compound to give amino.
Phase-1 reaction may not be able to produce complete inactive metabolite or sufficient hydrophilic metabolite, but makes the molecules amenable to undergo phase-II reactions easily. Thus Phase-I reactions increase hydrophilicity of molecule, reduces its stability and facilitates conjugation process.
Phase-2: Conjugation
Basic purpose of these reactions is to attach small, polar and ionizable endogenous compounds to the metabolite obtained from phase-I. This group includes glucoronic acid, sulfate, glycine and other amino acid so as to form water soluble conjugates. Parent compounds having -OH, -COOH, -NH2 functional groups can directly be conjugated by phase-II enzymes.
These highly hydrophilic metabolites excreted in urine are non-toxic and have no pharmacological effect. Other phase-II pathways like methylation, acetylation, glutathione conjugation (GSH) either terminate biological activity or protect body against chemically reactive metabolites.
Sites of Drug Biotransformation:
Liver is the main site of drug metabolism. All orally administered drugs pass through liver and hence are susceptible to first pass hepatic metabolism before reaching to systemic circulation. This decreases oral bioavailability of drugs like lidocaine, meperidine, propranolol, etc.
Intestine is also important for metabolism of some of the drugs (extra hepatic metabolism of xenobiotic) e.g. levodopa, chlorpromazine and diethyl stilbesterol. Enzymes like esterase and lipase present in intestinal wall; bacterial flora in intestinal and colon wall are helpful in metabolism. Other tissues like kidney, lungs, adrenal glands, brain and skin do take part in metabolism; but in a smaller extent.
Factors Influencing Drug Metabolism
- Genetic factors: Species difference must be considered when the data from animals are used to extrapolate the results in human.
- Physiological factors: Age (elderly disorder), individual variations in hormones, sex, physiological difference, nutritional status (diet), etc also affect rate of metabolism. If protein deficiency exists, then hepatic enzymes are also reduced. If lipid deficiency exists, rate of oxidative metabolism also decreases. If vitamins deficiency exists, activity of microsomal enzymes decreases.
- Pharmacodynamic factors: Dose, frequency of dose administration, route of administration, tissue distribution of drugs, proteins binding, etc. affect rate of metabolism.
- Environmental factors: The body can be exposed to environmental factors like air, water and food contaminants or pollutants by design or by accident. Pollutants like polychlorinated biphenyls are resistant to hydroxylation and thus may be hazardous to life as it remains there for long time after absorption.
Phase 1: Pathways Of Metabolism
Oxidative biotransformation: This is most preferred and important pathway of metabolism and follows following reaction scheme:
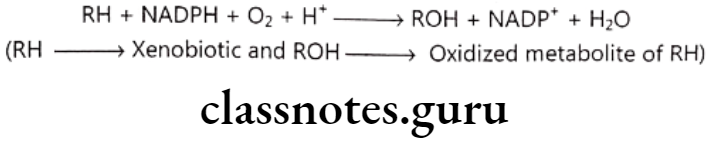
Enzyme carrying out this metabolism is mixed function oxidase (FMO) or mono- oxygenases. FMO is made up of many components like Cytochrome P-450 enzyme which transfers oxygen atom to RH, NADPH dependent cytochrome P-450 reductase, NADH linked cytochrome bs.
Cytochrome P-450 is a hemeprotein abundantly found in liver. Heme is the iron containing porphyrin i.e. protoporphyrin-IX and protein is apoprotein. Reduced form of cytochrome P-450 (i.e. Fe2+) binds with -CO to form a complex having absorption maximum at 450 nm, hence the family of enzyme is referred as cytochrome P-450.
Cytochrome P-450 metabolizes number of diverse substrates by various oxidative mechanisms. This enzyme is membrane bound and exists in different forms. The apoprotein portions of various cytochrome P-450 differ in their structure. Because the apoprotein portion is important in substrate binding and catalytic transfer of activated oxygen, specific form may oxidize specific substrate only.
Oxidative Reactions:
Oxidation of Aromatic Compounds:
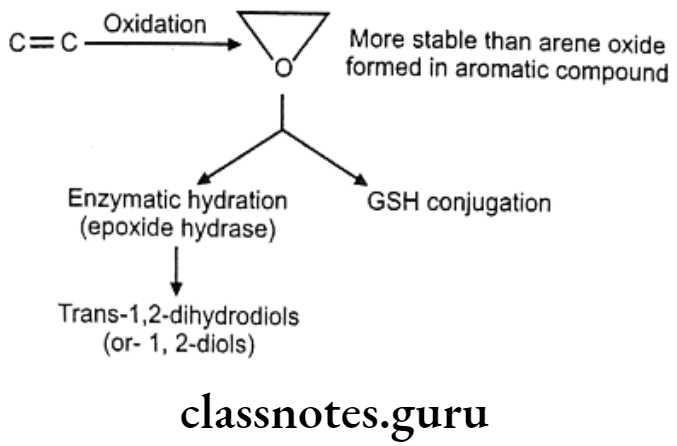
Hydroxylation: It causes oxidation of arene to arenol. Arene oxide (epoxide) is electrophilic, highly reactive because of strained 3-membered ring.
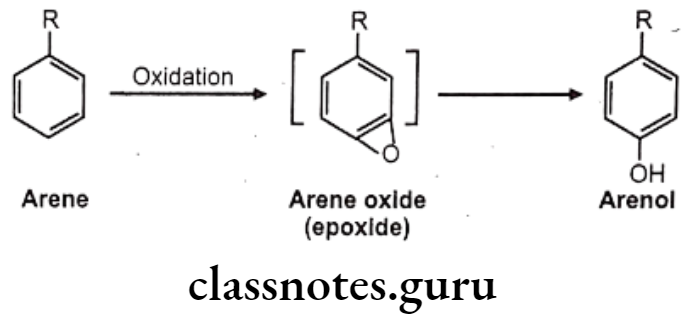
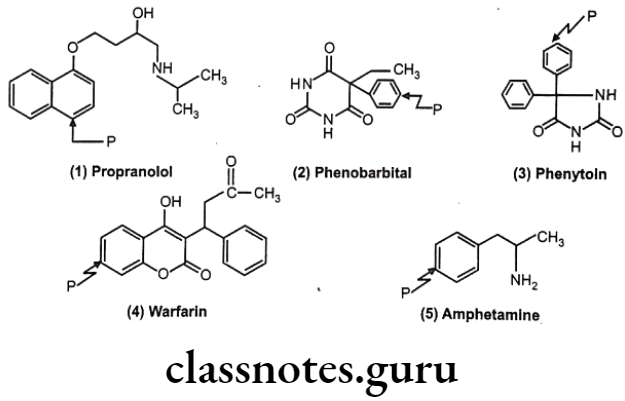
Many drugs follow this path e.g. (1) Propranolol, (2) Phenobarbital, (3) Phenytoin, (4) Warfarin, (5) Amphetamine.
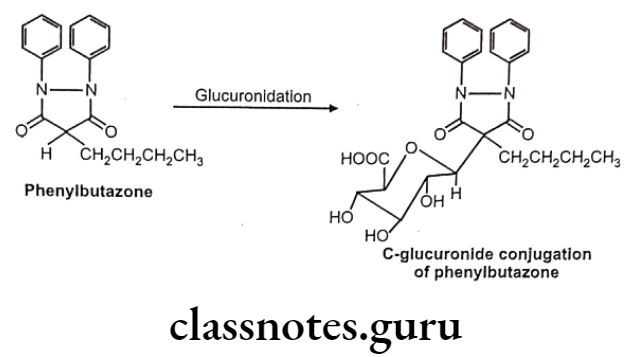
Sometimes the Phase-I hydroxyl metabolites are pharmacologically active. For example, in case of phenylbutazone, its metabolite is more active and hence it is termed as prodrug.
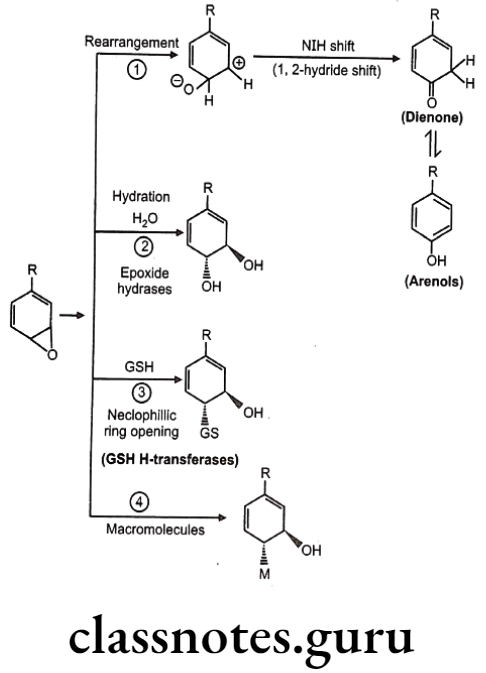
Detoxification of arene oxides: Arene oxides can be further inactivated by various ways as follows:
- Spontaneous rearrangement to arenols via NIH (1, 2-hydride) shift
- Hydration to trans-dihydrodiols
- Glutathione adducts
- Macromolecule adducts
Sometimes, in molecules like benzene, detoxification forms covalent adduct with nucleophillic groups present on DNA or RNA leading to cellular damage. Hence such molecules are very toxic to mammalian systems.
NIH Shift (1, 2-hydride shift): The novel intramolecular hydride migration is termed as NIH shift. It was named after National Institute of Health (NIH), USA, who discovered this process. Not all aromatic hydroxylation occurs through this process. The phenolic products of the hydroxylation of aromatics containing ortho-/para-directing substituents (F, Cl, Br, I, OH, NH2, CH3, CH2CH3, and OCH3) were primarily para-phenols.
In contrast, with aromatics containing meta-directing substituents (NO2 and CN), the phenolic products were a more even mixture of meta- and para-phenols. Ortho-fluorophenol was the only ortho-phenolic product observed. The nature of the products suggested that the reaction involves an enzyme-specific, electrophilic addition to the aromatic ring so as favour hydroxylation at either the meta- or para-positions.
With the fluoro-, chloro- and bromobenzene substrates, the values for the migration and retention of hydrogen during hydroxylation (NIH shift) were nearly identical when the hydrogen was either at the site of hydroxylation or at an adjacent site, indicating a possible common intermediate.
Metabolism of Phenytoin:
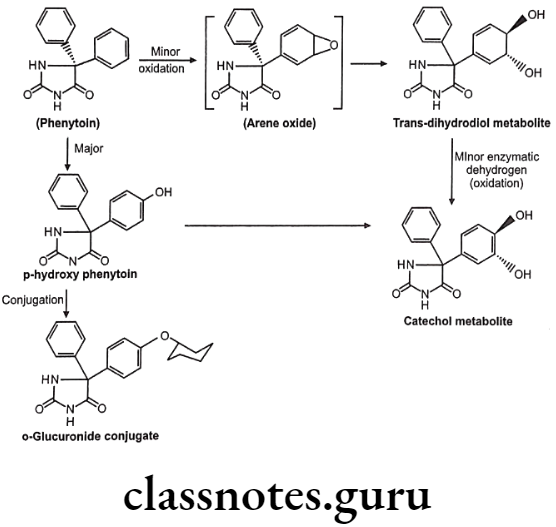
Oxidation of Olefins:
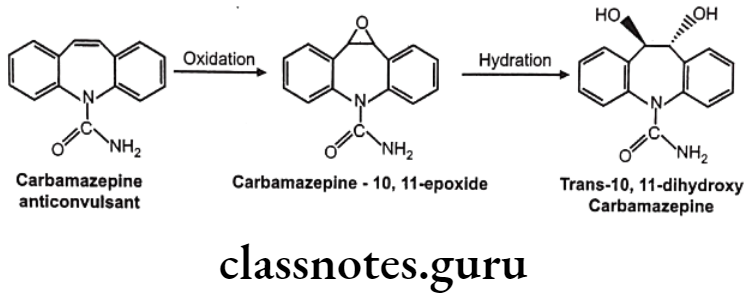
Olefinic epoxidation of carbamazepine:
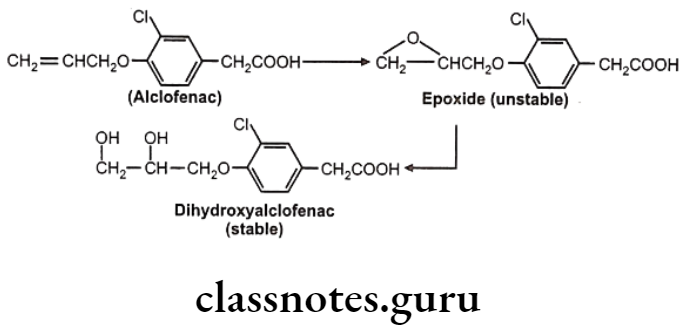
In drugs like alclofenac, secobarbital the epoxides are not stable and immediately converted to dihydroxy derivatives which are quite stable and isolable.
Sometimes epoxides formed are very reactive and produce toxic effects.
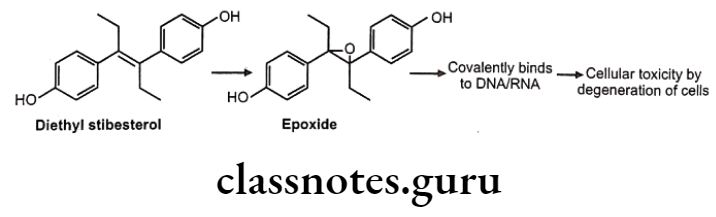
These reactive epoxides sometimes cause the destruction of cytochrome P450 enzymes e.g. epoxide of secobarbital, fluroxene (volatile anaesthetics) etc.
Oxidation of Benzylic C-Atoms:
Carbon attached to benzylic position in aromatic compound is susceptible to oxidation so as to form corresponding alcohol (carbinol) metabolite.
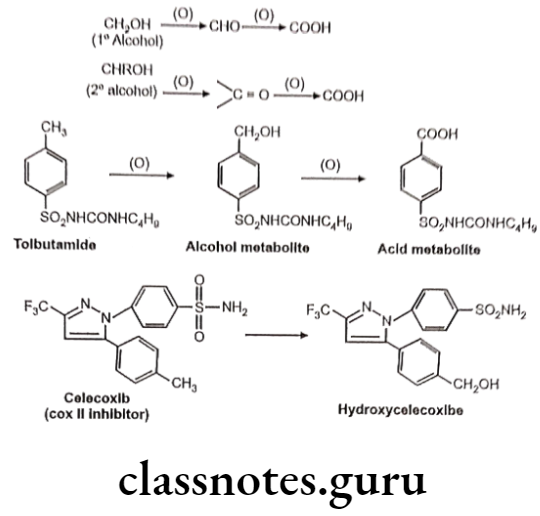
Oxidation of Aliphatic C-Atoms:
Metabolic oxidation at terminal methyl group is called as “o oxidation” and that of penultimate carbon atom (next to last carbon) is known as “o-1 oxidation”. This is generally observed in drug molecules with straight or branched alkyl chains.
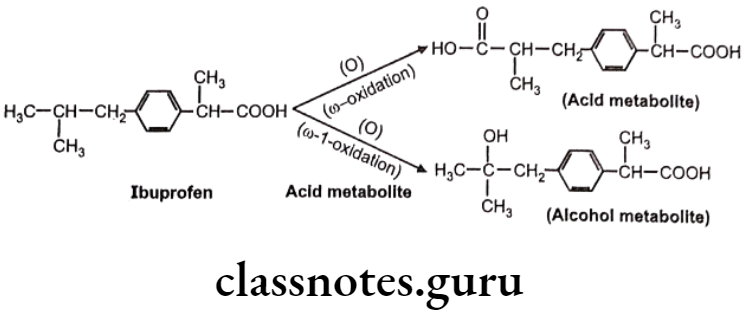
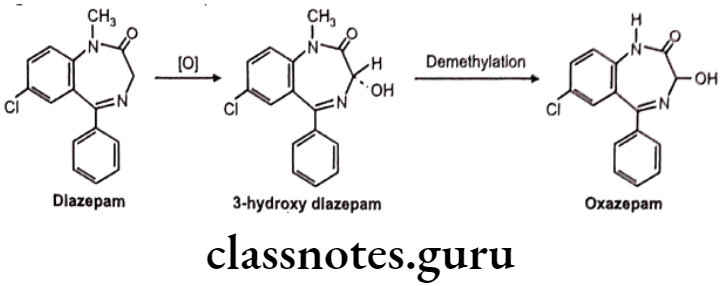
Oxidation of C-Atoms Alpha to Carbonyls and Imines:
e.g. Metabolism in diazepam.
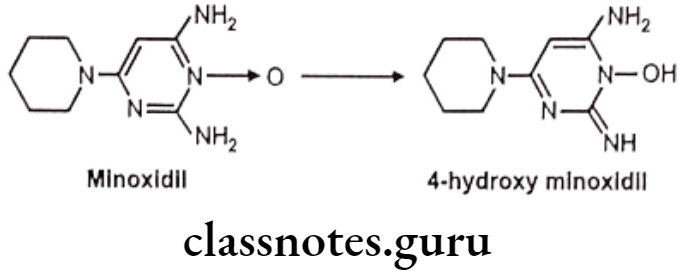
Oxidation of Alicyclic C-Atoms:
E.g. Oxidation of minoxidil
Oxidation of C-Hetero Atom Systems:
This involves two basic types of biotransformation processes. One hydroxylation of a-carbon attached directly to heteroatom leading to unstable intermediate and subsequent hydroxylation of heteroatom.
C-N Systems:
N-dealkylation: E.g. Codeine
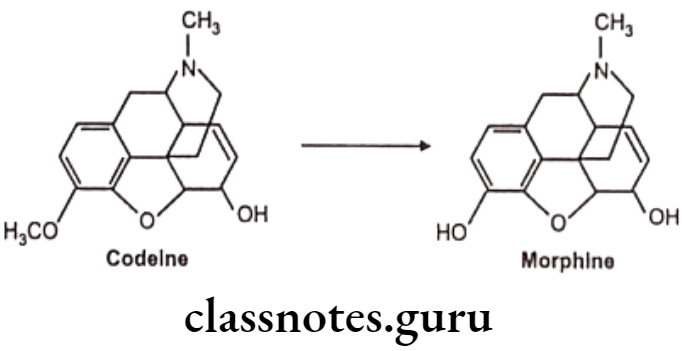
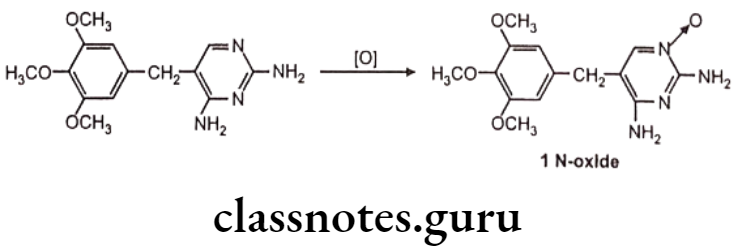
N-oxide formation: E.g. Trimethoprim
N-hydroxylation: Eg. Dapsone
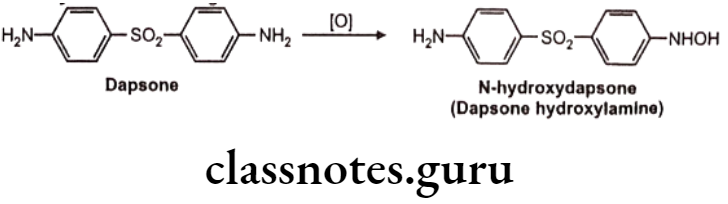
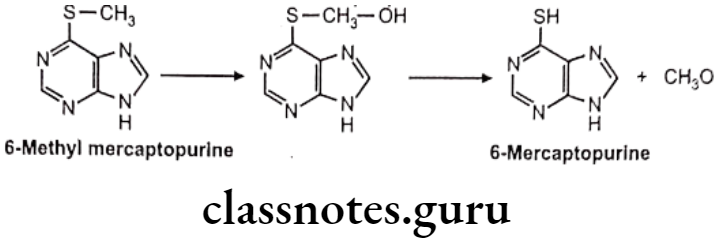
C-S Systems:
S-dealkylation: Eg. 6 methyl mercaptopurine
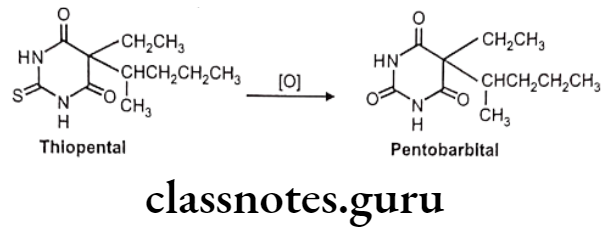
Desulfuration: Eg. Thiopental
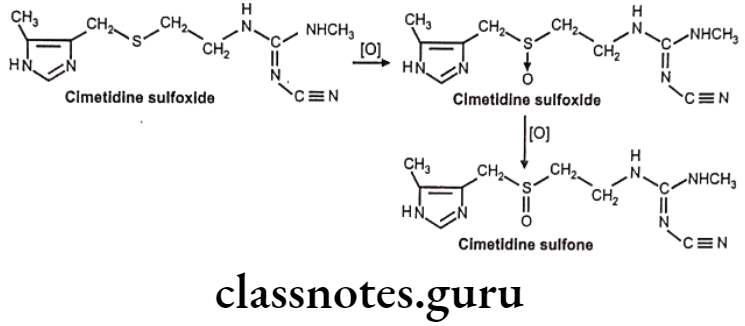
S-oxidation: E.g. Cimetidine
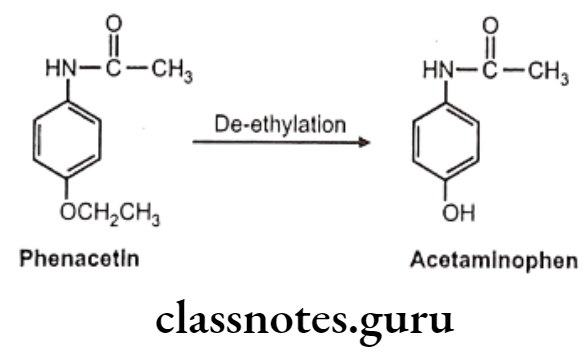
C-O Systems: E.g. Phenacetin
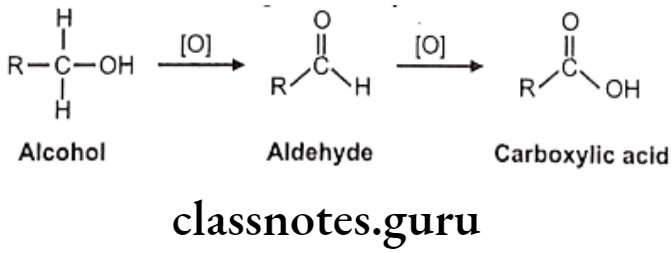
Oxidation of Alcohol and Carbonyl Functions:
Carbinol/alcohol metabolite intermediates obtained after oxidation of different hydroxylation processes which may undergo further oxidation to aldehydes or ketones and finally to acids. Secondary alcohol group is more polar and functionalized and hence may not get oxidized further, but it can get conjugated easily.
Miscellaneous: E.g. Chloroform
Reductive Reactions:
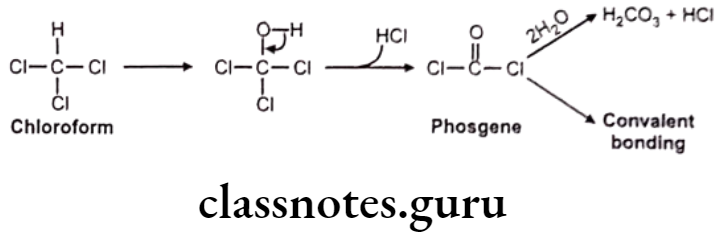
Reduction of Carbonyl Functions: Reduction of aldehydes E.g. Chloral hydrate.
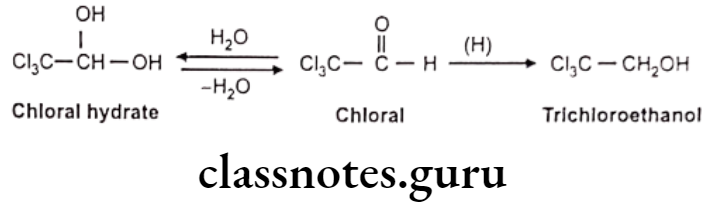
Reduction of Aromatic Ketone: E.g. Acetophenone
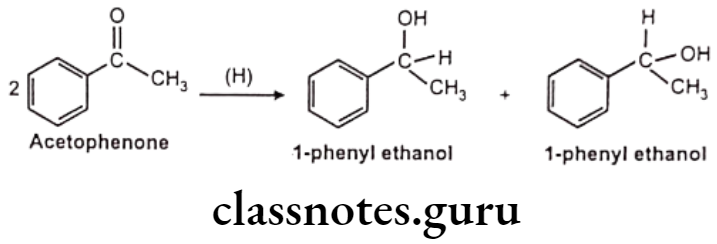
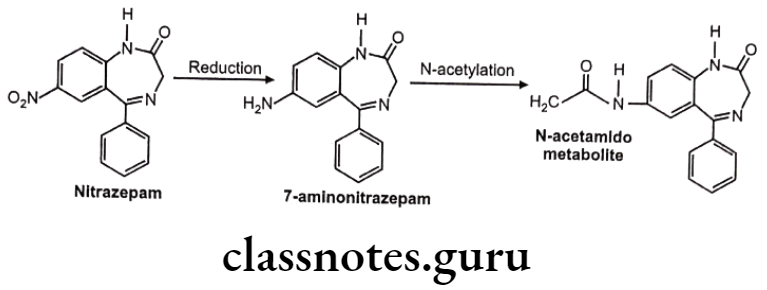
Reduction of N-Compounds: Eg. Nitrazepam.
Miscellaneous: E.g. Disulfiram
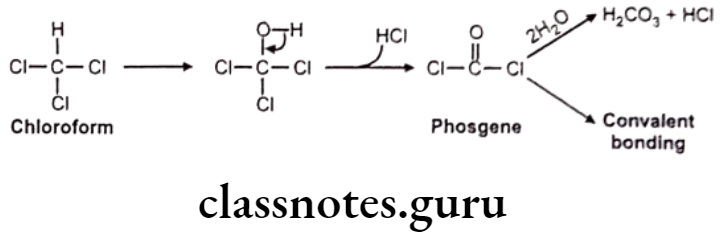
Hydrolytic Reactions:
Hydrolysis of esters and amides is done mainly by esterase, pseudo cholinesterase, amidases and deacylases. Esters are easy to hydrolyse as compared to amides. Functional groups like phosphate esters, sulphonyl urea, carbamate esters, epoxides and arene oxides are also susceptible to undergo hydrolysis.
Peptide hormones like insulin, prolactin etc. are also metabolized by hydrolysis. Sometimes ester hydrolysis may give pharmacologically active metabolite as in the case of chloramphenicol palmitate (prodrug). The palmitate ester is used to minimize the bitter test of the drug and increase palatability in the paeidatric patients.
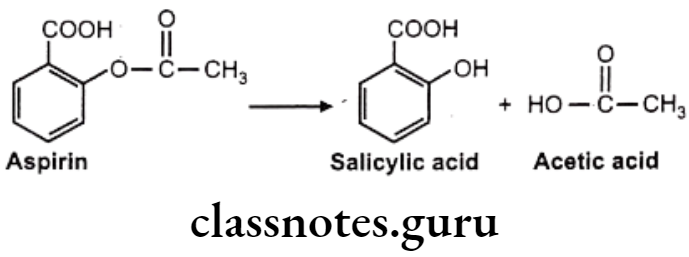
Hydrolysis of Esters: E.g. Aspirin
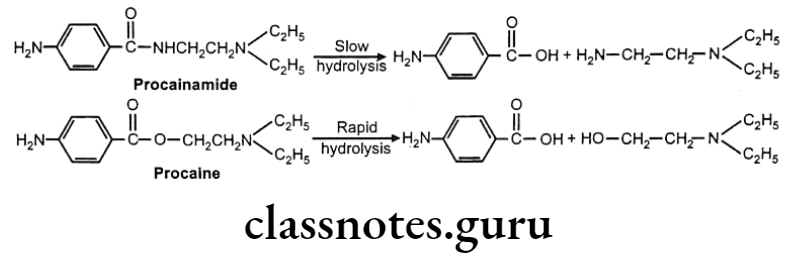
Hydrolysis of Amides: E.g. Procainamide
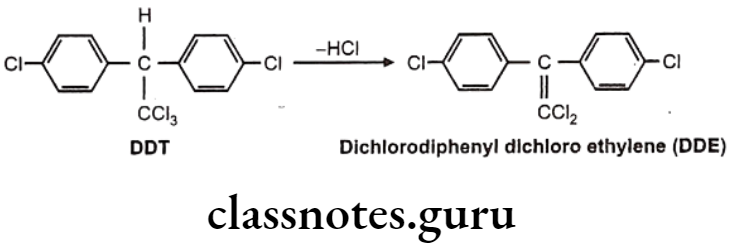
Hydrolytic Dehalogenation: E.g. Dichloro diphenyls trichloroethane
Phase-2: Conjugation Reactions
Phase-1 metabolites need further conversion for easy excretion and hence small, polar and ionizable endogenous molecules like glucuronic acid, sulfate glycine and glutamine are attached to them leading to generation of Phase-II metabolites. These are biologically inactive, non-toxic. Some other phase-II reactions like methylation and acetylation do not increase water solubility, but terminate pharmacological activity. True detoxification occurs in phase-II pathway. Glutathione (GSH) combines with reactive phase-I metabolites and prevent them combining with biomolecules like DNA/RNA, thus cytotoxicity is prevented.
Mechanism of Action: Conjugating groups are activated in the forms of coenzymes and then are attached / transferred to accepting substrates; while in some other cases like glycine and glutamine conjugation, substrate is activated rather than conjugating groups. Transferase enzyme plays an important role in activation.
Glucuronic Acid Conjugation:
This is the most common pathway because:
- D-glucuronic acid is easily available (obtained from D-glucose).
- Many functional groups can combine enzymatically with glucuronic acid.
- Glucuronyl moiety can ionize carboxylate and polar hydroxyl group to increase water solubility.
Mechanism of Action: B-glucuronide moiety is attached to substrate with the help of microsomal uridine-5-diphospho-glucuronosyl transferase enzyme. All glucuronide conjugations have B-linkage or B-conjugation at C1.. Many functional groups undergo glucuronidation. According to hetero atom (O, S, N) attached to C1 of glucuronyl moiety, metabolic products of these conjugation are classified as –
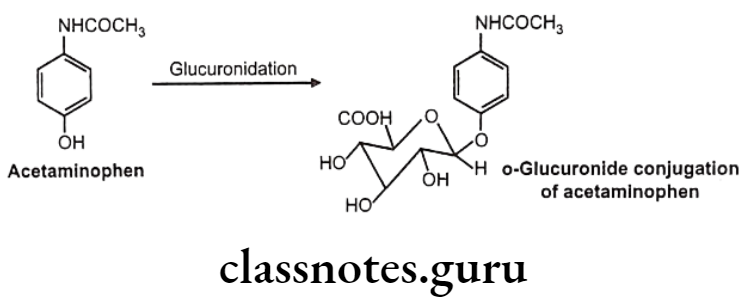
- Oxygen glucuronidee.g.: Acetaminophen
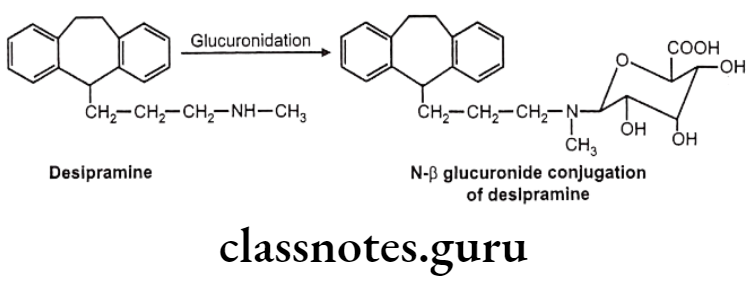
- Nitrogen glucuronides – e.g.: Desipramine
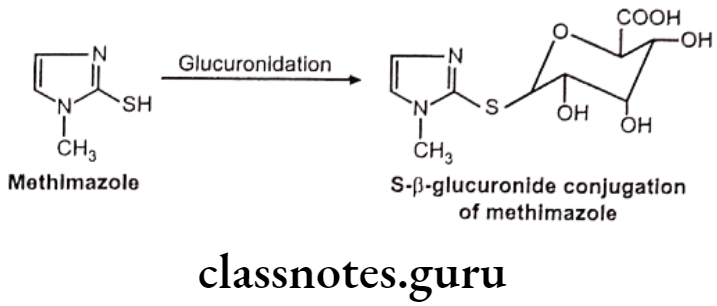
- Sulfur glucuronides – e.g. Methimazole
- Carbon glucuronides – e.g. : Phenylbutazone
O-Glucuronides
N-glucuronides:
These are formed with aromatic amines, aliphatic amines, amides and sulfonamides. But generally aliphatic and aromatic amines are not glucurylated. They undergo N-acetyl preferably.
S-glucuronides: This is observed very rarely.
C-glucuronides:
Apart from xenobiotic, many endogenous substrates like bilirubin, steroidal hormones are also eliminated as glucuronide conjugates. In neonates and infants glucuronidation processes are not fully developed. So in such cases serious toxicity may be seen because of accumulation of drugs like bilirubin.
e.g. Neonatal hyperbilirubinemia- Inability to conjugate bilirubin with ẞ-glucuronide.
Gray baby syndrome – Inability to conjugate chloramphenicol with glucuronide.
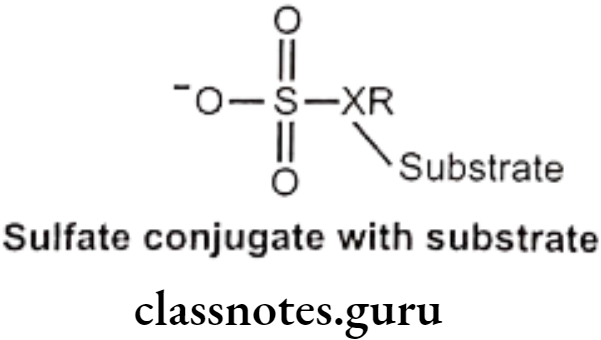
Sulfate Conjugation:
This occurs primarily in phenols and also in alcohols, aromatic amines etc. Amount of sulfate available for conjugation is limited. Endogenous substance like heparin, steroids, catecholamine and thyroxin are also sulfate conjugated.
Mechanism: Sulfate group from PAPS (3-phosphoadenosine-5-phosphosulfate) is transferred to substrate with the help of sulfotransferase enzymes (in liver, kidney, intestine etc.) Phenols are more susceptible to this conjugation.
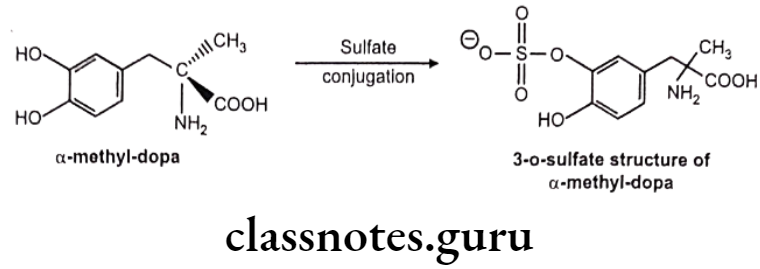
Examples:
a-Methyl dopa:
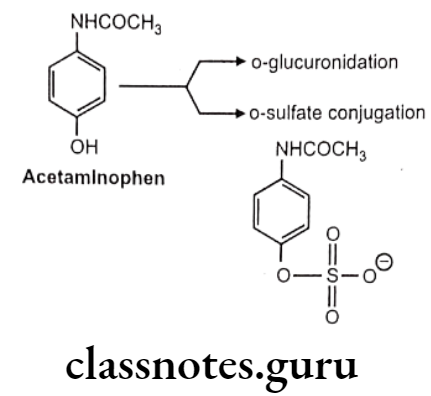
Acetaminophen:
Conjugation with Glycin, Glutamine and Other Amino Acids
This is specifically used for conjugation of aromatic acid and aryl alkyl acids. Limited quantities of amino acids are available for conjugation.
Mechanism: Substrate is activated with ATP and CoA to form Acyl-CoA complex. This acylates glycine/ glutamine under influence of specific enzymes like glycine/ glutamate N-acyltransferase Activation and acylation take place in mitochondria of liver/kidney cells.
Glycin conjugation:
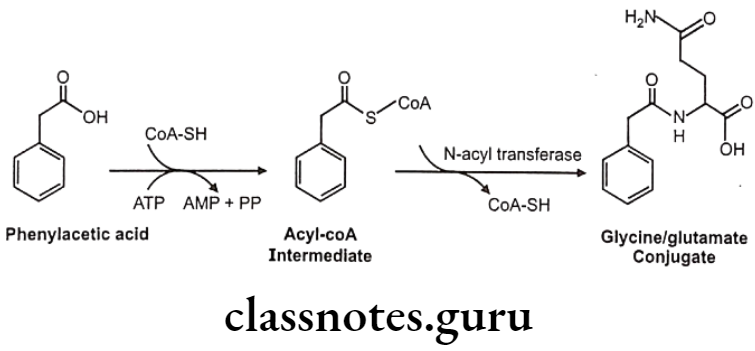
Glutamine conjugation:
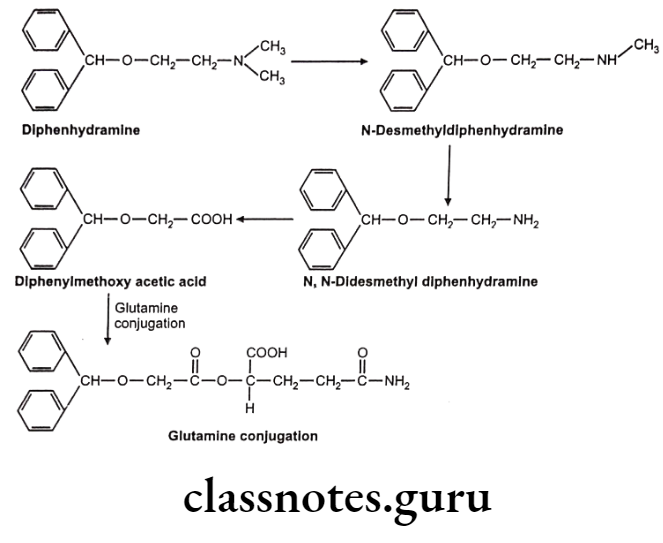
Glutathione (GSH) or Mercapturic Acid Conjugates:
This is also an important pathway for detoxification of chemically reactive electrophilic compound. GSH protects vital cellular constituents against chemically reactive species by its nucleophilic -SH group. This SH group reacts with electron deficient compound to form S-substituted GSH adduct. GSH conjugated with xenobiotic is further biotransformed into mercapturic acid, involving enzymatic cleavage of 2 amino acids (glutamic and glycine). GSH conjugation does not involve initial formation of activated coenzymes of substrate.
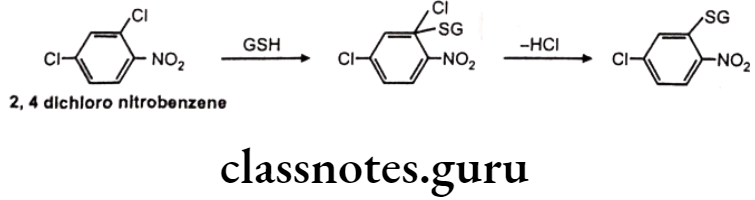
Mechanism of Action: Nucleophilic displacement at an electron deficient carbon/ heteroatom or nucleophilic addition to an electron deficient C=C. Many industrial chemicals are detoxified by this route.
Example: 2,4-dichloro nitrobenzene
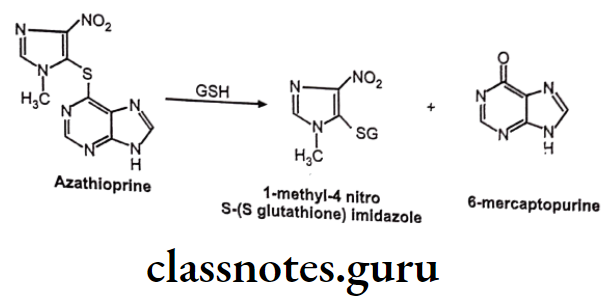
GSH Conjugation Involving Substitution At Heteroatom Like N, O, S:
Example: Azathioprine
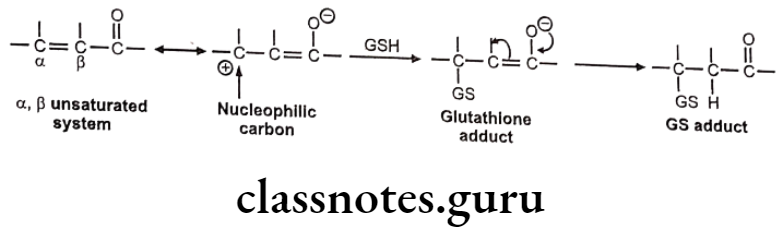
Nucleophilic Addition Of GSH To Electron Deficient C=C:
The C=C is generally made electron deficient by resonance or conjugation with C=O group, esters, amides, nitriles etc. It generally occurs in a, B-unsaturated systems and undergoes Michael addition reaction with GSH to give GSH adduct.
Mechanism of nucleophilic addition of GSH to electron deficient C=C is as follows:
Example: Ethacrynic acid (Diuretic)
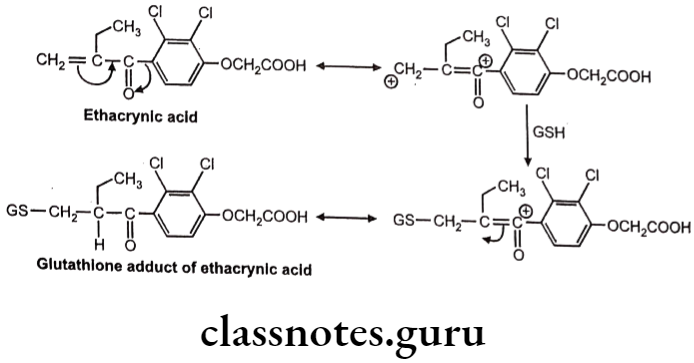
Sometimes chemically reactive a,ẞ-unsaturated systems are generated during various biotransformations. GSH reacts with these systems by Michael addition reaction.
Example: Acetaminophen
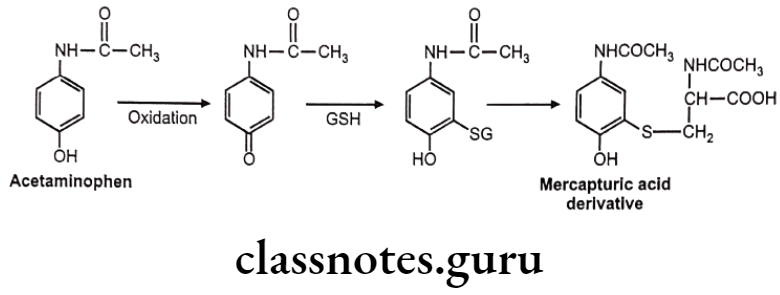
GSH conjugate may also cause toxicity due to metabolic intermediates which are electrophilic. Such electrophiles attack on DNA or RNA and produce cytotoxicity.
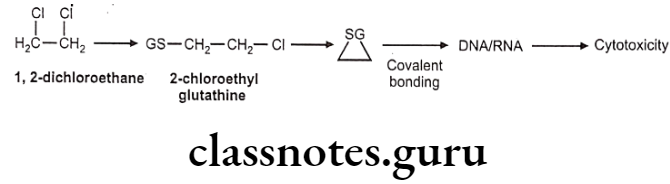
Example: 1,2-dichloroethane
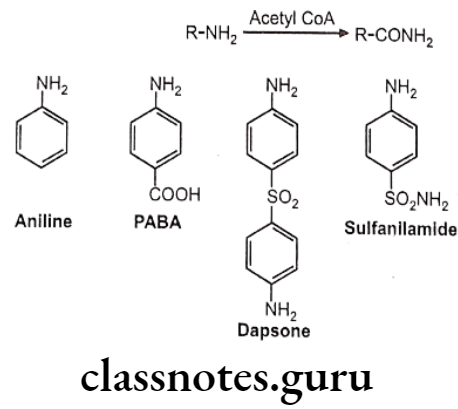
Acetylation:
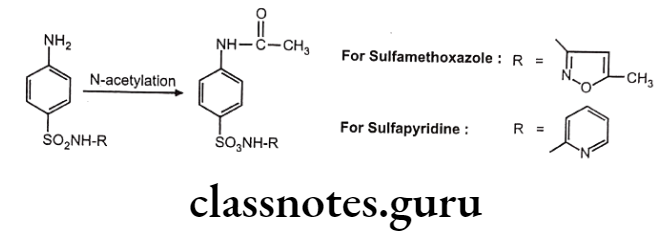
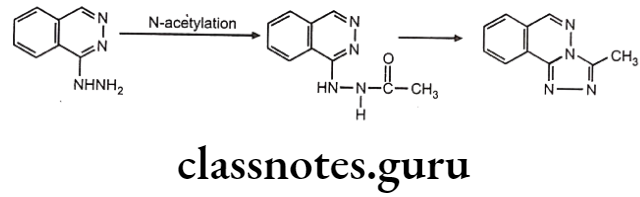
Drugs containing primary amines undergo metabolism by this route. It includes various functional groups like aromatic amines, sulphonamides, hydrazines, hydrazides and primary aliphatic amines. Soluble N-acetyl transferase are the enzymes which transfer the acetyl group from acetyl CoA to the accepting amino substrate. N-acetylation process doesn’t affect water solubility, but terminates the biological activity.
Examples:
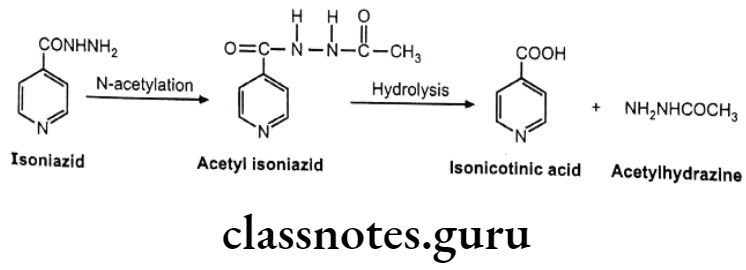
Isoniazid
Nitrazepam
Biotransformation Of Sulfonamide:
Biotransformation Of Hydrazine And Hydride Derivative:
Acetylation Polymorphism:
The acetylation pattern of many drugs in human being can be classified as-slow and rapid acetylation. i.e. drug conjugates with acetyl CoA slowly or rapidly. This phenomenon is called as acetylation polymorphism and individual are called as slow acetylators or rapid acetylators. This variation in acetylation is genetic and depends on activity of N-acetyltransferases.
Asians and Eskimos are rapid acetylators and hence produce insufficient response at therapeutic dose. Egyptians and western population are slow acetylators and may produce adverse reaction at given dose. e.g. Half-life of isoniazid in rapid acetylators is 45-80 minutes, while in slow acetylators it is 140-200 minutes.
In the slow acetylators, accumulation of drug is seen which gives higher response and higher side effects. Slow acetylators are also susceptible to drug interaction. e.g. In isoniazid slow acetylators, isoniazid inhibit metabolism of phenytoin, thus its accumulation is more leading to toxicity.
While in case of rapid acetylators, the acetyl hydrazine metabolite formed by rapid biotransformation of isoniazid undergoes N-oxidation mediated by cytochrome P450 and gives reactive intermediate which may covalently bind to liver cells. So rapid acetylators have a greater risk of liver injury.
Methylation:
Catechols, phenols, amines, N-heterocyclic and thiol compounds undergo methylation. It plays an important role in inactivation of many physiologically active biogenic amines like dopamine, 5-HT, histamine and norepinephrine. It is minor pathway of conjugation. Most methylated products are inactive and non-toxic.
Coenzyme like 5-adenosylmethione and enzymes like methyl-transferase play crucial role here. Methyl-transferase is a group of enzymes which contain catechol-o-methyltransferase (COMT), phenol-o-methyl-transferase, non-specific N-methyl-transferase and S-methyl-transferase etc.
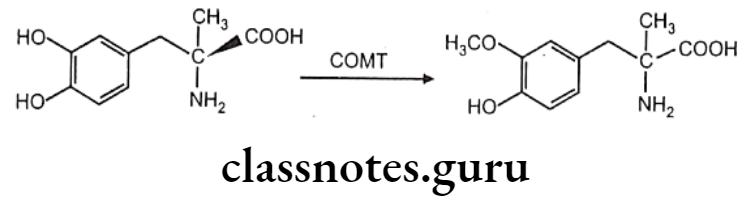
COMT causes O-methylation of neurotransmitter like norepinephrine, dopamine. Other drugs methylated by COMT include methyl dopa. Bismethylation do not occur in above case. 1,2-dihydroxy group. is necessary for the action of COMT. Hence in case of 1,3 and 1,4-dihydroxy benzene scaffold selective O-methylation is not possible. Hence isoproterenol undergoes O-methylation, whereas terbutaline does not.
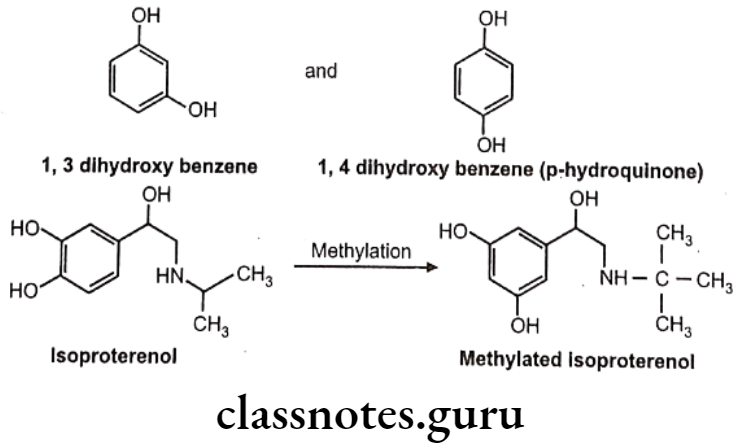
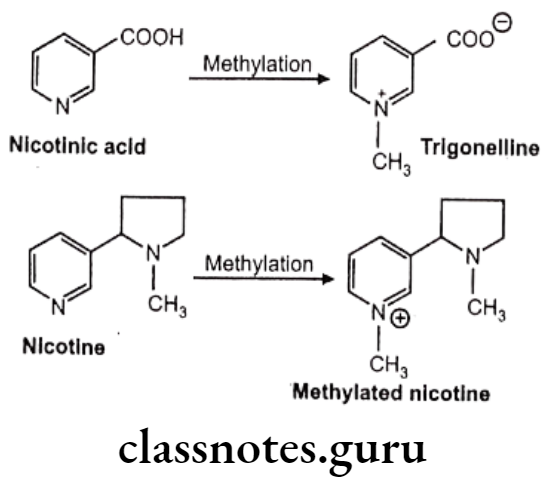
Other examples:
Factors Affecting Drug Metabolism
- Age Difference
- Species and Strain
- Genetic/Hereditary factors.
- Sex differences
- Enzyme Induction
- Enzyme Inhibition
Age Difference:
In case of fetus and newborn babies, oxidative and conjugative enzymes are either deficient or underdeveloped. Hence their response to metabolism of certain drug is very different. e.g. Oxidative metabolism of tolbutamide is less in newborn, while half-life in adult is 8 hours and in infants it is greater than 40 hrs.
Activity of glucuronyltransferase is less in infants. Neonatal hyperbilirubinemia and gray baby syndrome are examples of such underdeveloped metabolism systems, in which infants are unable to glucuronidate bilirubin and chloramphenicol respectively.
Species And Strain:
Metabolism of many drugs and foreign compounds is species dependent. Cats lack glucuronyltransferase enzymes, hence phenolic xenobiotic are metabolized by sulfation, rather than glucuronidation only. But in pigs, due to lack of sulfotransferase enzymes, these phenolic drugs are metabolized by glucuronidation only. Example of strain difference includes cottontail rabbit liver microsomes metabolizes hexobarbital more rapidly than New Zealand rabbit liver microsomes.
Genetic/Hereditary Factors:
As seen above, acetylation of isoniazid is different in different races and accordingly they are categorized as – rapid and slow acetylators. Genetic factor influences rate of oxidation of some drugs like phenytoin, phenylbutazone etc.
Sex Differences:
Nicotine and aspirin are metabolized differently in women and men. Adult male rats metabolize many foreign compounds at faster rate than female.
e.g. N-demethylation of aminopyrine.
Enzyme Induction:
Activity of cytochrome P-450 dependent Mixed Function Oxidases (MFO) can be enhanced upon exposure to many drugs, pesticides, polycyclic aromatic hydrocarbons etc. This is called as enzyme induction. Enzyme induction causes increase in drug metabolism and decreases duration of drug action. e.g. Induction of microsomal enzyme by phenobarbital increases metabolism of warfarin and thus decreases anticoagulant effect.
Other example is enhanced metabolism of endogenous substances like cortisol, testosterone, vitamin D and bilirubin by phenobarbital. Phenobarbital also induces glucuronyltransferase enzyme and thus increases conjugation of bilirubin with glucuronic acid and hence it can be used to treat hyper bilirubinemia in neonates.
Enzyme Inhibition:
Some drugs can inhibit metabolism of other drugs. Decreased metabolism leads to accumulation, prolonged drug action and serious adverse effect. Enzyme inhibition takes place by many mechanisms like substrate competition, interference with protein synthesis, fractionation of drug metabolizing enzymes and hepatotoxicity leading to impairment of enzyme activity etc. e.g. Phenylbutazone inhibits metabolism of S (-) warfarin stereo- selectivity due to which, increased anticoagulant effect (hypoprotothombinemia) is seen. Other examples include metabolism of phenytoin is inhibited by drugs like disulfiram, isoniazid and chloramphenicol.
Miscellaneous Factors:
- Dietary factor – Protein or carbohydrate ratio affects metabolism of many drugs. Vitamins, minerals, starvation and malnutrition also influence drug metabolism.
- Physiological factor Pathogenic state of liver, pregnancy, hormonal disturbances, etc. also affect metabolism.
e.g. Indoles present in vegetables like cabbage, cauliflower and polycyclic aromatic hydrocarbons present in boiled beef causes enzymes induction and increases metabolism of many drugs.
Stereochemical Aspects Of Drug Metabolism
Stereoselectivity is also observed in drug disposition particularly for those processes which depend on an interaction with a chiral biological macromolecule, e.g. active transport processes, binding to plasma proteins, and drug metabolism. In drug metabolism, stereoselectivity in metabolism is probably responsible for the majority of the differences observed in enantioselective drug disposition.
Stereoselectivity in metabolism may arise from differences in the binding of enantiomeric substrates to the enzyme active site and/or be associated with catalysis owing to differential reactivity and orientation of the target groups to the catalytic site. As a result, pair of enantiomers is frequently metabolized at different rates and/or via different routes to yield alternative products.
Example: Drug receptor interaction of (R)-(-)-epinephrine, (S)-(+)-epinephrine and N-methyl dopamine.
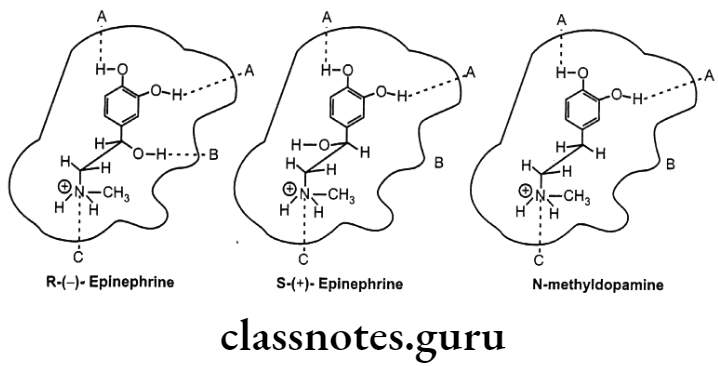
The stereoselectivity of the reactions of drug metabolism may be classified into three groups in terms of their selectivity with respect to the substrate, the product, or both. Thus, there may be substrate selectivity when one enantiomer is metabolized more rapidly than the other; product stereoselectivity, in which one particular stereoisomer of a metabolite is produced preferentially; or a combination of the above, i.e. substrate- product stereo- selectivity, where one enantiomer is preferentially metabolized to yield a particular diastereoisomeric product.
An alternative classification involves the stereochemical consequences of the transformation reaction. Using this approach, metabolic pathways may be divided into five groups:
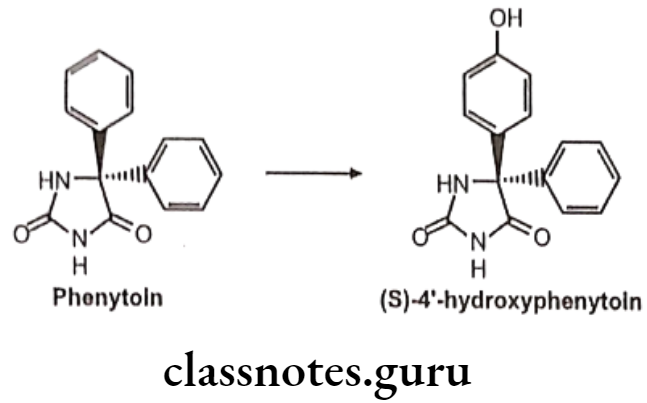
Prochiral To Diastereoisomer Transformations: Metabolism taking place either at a prochiral center or on an enantiotopic group within the molecule e.g. phenytoin undergoes stereoselective para-hydroxylation to yield (S)-4′-hydroxyphenytoin in greater than 90% enantiomeric excess.
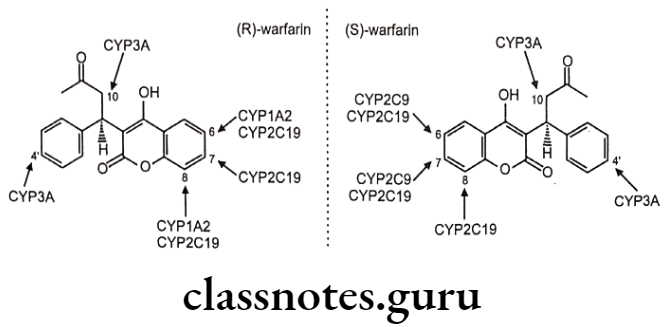
Chiral To Diastereoisomer Transformations: The individual enantiomers of a drug undergo metabolism at a site remote from the center of chirality with no configurational consequences e.g. (S)-warfarin undergoes aromatic oxidation mediated by CYP 2C9 in the 7- and 6-positions to yield (S)-7-hydroxy- and (S)-6-hydroxywarfarin in the ratio 3.5:1.
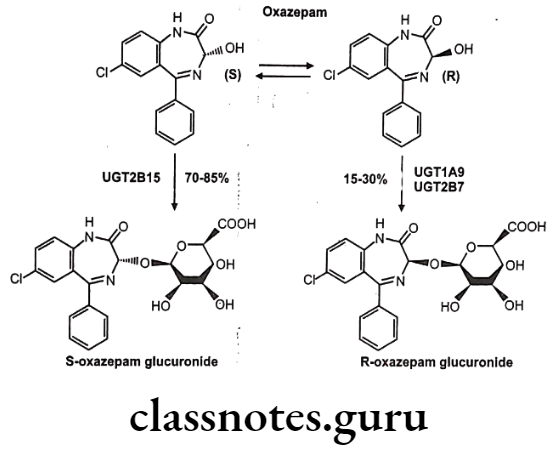
Chiral To Diastereoisomer Transformations: A second chiral center is introduced into the drug either by reaction at a prochiral center or via conjugation with a chiral conjugating agent e.g. stereoselective glucuronidation of oxazepam.
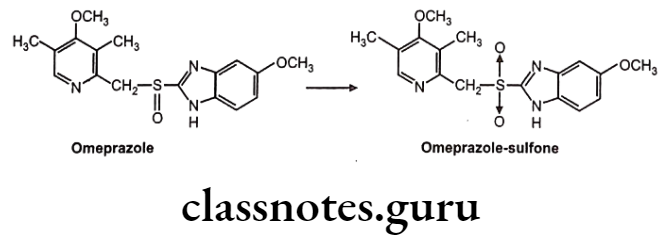
Chiral To Achiral Transformations: The substrate undergoes metabolism at the center of chirality, resulting in a loss of asymmetry e.g. oxidation of the benzimidazole proton pump inhibitors, e.g., Omeprazole, which undergoes CYP 3A4-mediated oxidation at the chiral sulfoxide to yield the corresponding sulfone. The reaction shows tenfold selectivity for the S-enantiomer in terms of intrinsic clearance.
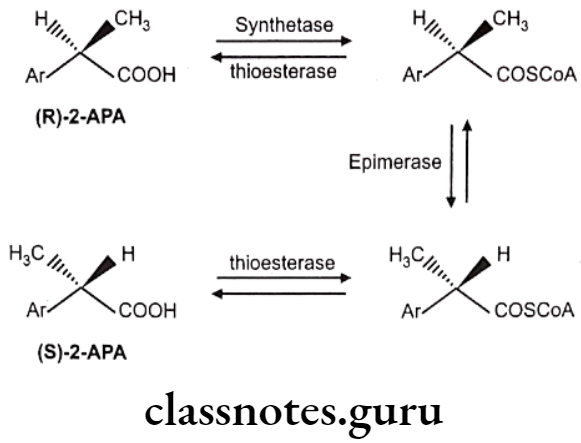
Chiral Inversion: One stereoisomer is metabolically converted into its enantiomer with no other alteration in structure, e.g. 2-arylpropionic acids like ibuprofen, fenoprofen, flurbiprofen, ketoprofen, and the related 2-aryloxypropionic acid herbicides, e.g., haloxyfop. The reaction is essentially stereospecific, the less active, or inactive, R-enantiomers undergoing inversion to the active S-enantiomers.
Multiple Choice Questions
Question 1. The term “biotransformation” includes the following:
- Accumulation of substances in a fat tissue
- Binding of substances with plasma proteins
- Accumulation of substances in a tissue
- Process of physicochemical and biochemical alteration of a drug in the body
Answer. 4. Process of physicochemical and biochemical alteration of a drug in the body
Question 2. Biotransformation of the drugs is to render them:
- Less ionized
- More pharmacologically active
- More lipid soluble
- Less lipid soluble
Answer. 4. Less lipid soluble
Question 3. Metabolic transformation (phase 1) is:
- Acetylation and methylation of substances
- Transformation of substances due to oxidation, reduction or hydrolysis
- Glucuronide formation
- Binding to plasma proteins
Answer. 2. Transformation of substances due to oxidation, reduction or hydrolysis
Question 4. Confirmational isomerism is
- Cis-trans isomerism
- Optical isomerism
- Dextro and levo rotatory
- Non-identical spatial arrangement of the atoms in molecule resulting from rotation about one or more simple bonds
Answer. 4. Non-identical spatial arrangement of the atoms in molecule resulting from rotation about one or more simple bonds
Question 5. Conjugation is:
- Process of drug reduction by special enzymes
- Process of drug oxidation by special oxidases
- Coupling of a drug with an endogenous substrate
- Solubilization in lipids
Answer. 3. Coupling of a drug with an endogenous substrate
Question 6. Which of the following processes proceeds in the second phase of biotransformation?
- Acetylation
- Reduction
- Oxidation
- Hydrolysis
Answer. 1. Acetylation
Question 7. According to pH partition theory, a weakly acidic drug will most likely be absorbed from the stomach because the drugs which exist primarily in the:
- Un-ionized, more lipid soluble
- Ionized, more water soluble
- Form a weak acid and more soluble in water media
- Ionic form of the drug which facilitates diffusion
Answer. 1. Un-ionized, more lipid soluble
Question 8. Conjugation of a drug includes the following EXCEPT:
- Glucuronidation
- Sulphate formation
- Hydrolysis
- Methylation
Answer. 3. Hydrolysis
Question 9. Metabolic transformation and conjugation usually results in an increase of a substance’s biological activity:
- True
- False
Answer. 2. False
Question 10. An antagonist is a substance that:
- binds to the receptors and initiates changes in cell function, producing maximal effect.
- binds to the receptors and initiates changes in cell function, producing submaximal effect.
- interacts with plasma proteins and doesn’t produce any effect.
- binds to the receptors without directly altering their functions.
Answer. 4. binds to the receptors without directly altering their functions.
Question 11. Irreversible interaction of an antagonist with a receptor is due to:
- Ionic bonds
- Hydrogen bonds
- Covalent bonds
- All of the above
Answer. 3.Covalent bonds