
Chapter 5 Drugs Action On Central Nervous System
General Anesthetics
Introduction
Earlier the pain-producing surgical procedures and dental surgeries were undertaken without the aid of acceptable anesthetic agents. Various chemical methods were adopted at that time included intoxication with ethanol, hashish, or opium, whereas physical methods included packing a limb in ice, creating ischemic conditions with tourniquets, inducing unconsciousness by a blow to the head, or the most common technique, employing strong armed assistants to hold down the helpless patient during the entire surgical procedure.
Nitrous oxide also known as “laughing gas” was first time used by Hartford dentist Horace Wells as a surgical anesthetic in 1844. This gas is still commonly used today, especially in combination with other anesthetic and analgesic agents. Another agent cyclopropane was popularly used as general anesthetics but because of its explosive nature like diethyl ether it is no longer used. For many years, the inhalational anesthesia was used for all major surgical procedures, however recently intravenous anesthesia has become more commonly used technique.
The inhalational anesthetic agents used today are hydrocarbons and ethers with halogen like, Cl, Br, or F and intravenous anesthetics (short acting barbiturates e.g. Thiopental sodium) possess most of the ideal characteristics.
Ideal Characteristics General Anesthetics
The ideal anesthetic state is characterized by a loss of all sensations and includes analgesia and muscle relaxation. It should possess following characteristics, but currently there is no such ideal agent that fulfils all these characteristics.
- It should be non-reactive.
- It should be inexpensive.
- It should be non-toxic.
- It should be non-flammable / non-explosive.
- It should produce rapid and pleasant induction of surgical anesthesia.
- It should produce rapid and pleasant withdrawal from anesthesia.
- It should produce adequate relaxation of skeletal muscles.
- It should be potent enough to permit adequate oxygen supply in mixture.
- It should have wide margin of safety.
- It should be free of adverse effects.
- It should be chemically compatible with anesthetic devices.
General anesthesia is the induction of a state of unconsciousness with the absence of pain sensation over the entire body, through the administration of Anesthetic drugs. General anesthetic drugs produce controlled, reversible depression of the functional activities of the CNS producing loss of sensation and consciousness.
Purpose of General Anesthesia
- To get relief of pain (analgesia).
- To block memory of the procedure (amnesia).
- To produce unconsciousness.
- To inhibit normal body reflexes to make surgery safe and easier to perform.
- To relax the muscles of the body.
Mechanism of Action of General Anesthetic Agents
General anesthetic agents are positive modulators of the action of GABA on GABAA receptors by binding to allosteric binding sites. They act by facilitation of GABA receptors to promote chloride ion conductance and at therapeutic concentrations, some of the agents (e.g. ethanol and phenobarbital) depress the function of ionotropic glutamate receptors (excitatory), which may contribute to the overall anesthetic effect.
Stages of Anesthesia
After administration of general anesthetic agent (lipophilic and unionised) to a patient, it passes most rapidly into the central nervous system. As the blood concentration of the agent increases, penetration into the CNS increases which leads to increased depth of anesthesia. Guedel in 1937, has defined four stages of anesthesia as follows:
- Stage-1 (Stage of analgesia): This stage is the period from the beginning of administration of anesthesia to the maintain consciousness. Analgesia is produced and the patient progressively loses pain, therefore this stage is also called stage of analgesia. Since, in this stage higher cortical centers are depressed. This stage is also known as cortical stage
- Stage-2 (Stage of delirium or stage of excitement): This stage extends from the loss of consciousness through a stage of irregular and specific breathing to the re-establishment of regular breathing. There is loss of consciousness. Due to further depression of cortical centers, patient leads to the excitement and may shout or struggle violently. Hence this stage is called as the stage of excitement. In this stage patient may salivate, vomit or develop cough excessively. In this stage the respiration is normal and regular.
- Stage-3 (Stage of surgical anesthesia): In this stage excitement is lost and skeletal muscles are relaxed and hence most of the operative procedures are performed in this stage. This stage is divided into four different planes with progressive increase in depth of anesthesia and decrease in respiration and eye movement.
- Stage-4 (Stage of medullary paralysis or respiratory paralysis): The respiration ceases as the depth of anesthesia reaches to stage-IV. This may happen due to the overdose or toxic effect of the anesthetic. In this stage respiratory and cardiovascular system gets collapsed and the tissue rapidly becomes anoxic. There is no any eye movement.
To avoid the toxic effect of anesthesia, anesthetics are either administered by intravenous or rectal rout (basal anesthetics or fixed anesthetics), which lead to loss of consciousness before volatile anesthetics given and hence transition from complete consciousness to the surgical anesthesia will be rapid and safe.
Pre-anesthetic Medication
For the safe and effective response of general anesthetics, pre-anesthetic medications are generally recommended. These are:
- Hypnotics: Can be given one night before surgery, to assure good night sleep.
- Atropine (or hyoscine): Can be given two hours before surgery to prevent excess secretion of saliva or mucous which might obstruct the process is anesthesia.
- Morphine (or pethidine): It is given to minimize fear and anxiety.
Classification Of General Anestnetics
- Inhalation anesthetics: Halothane, Methoxyflurane, Enflurane, Sevoflurane, Isoflurane, Desflurane.
- Ultra short-acting barbitutrates: Methohexital sodium, Thiamylal sodium, Thiopental sodium.
- Dissociative anesthetics: Ketamine hydrochloride.
Inhalation Anesthetics
Halothane:
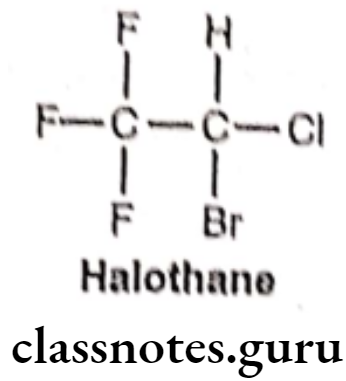
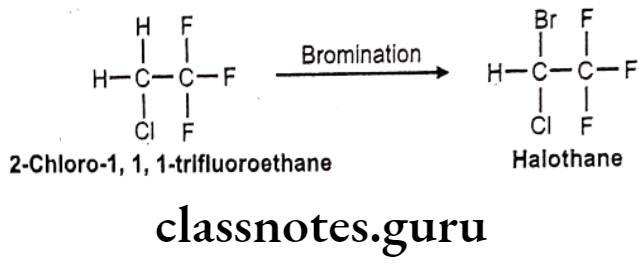
- Halothane is chemically, 2-bromo-2-chloro-1,1,1-trifluoroethane.
- It was introduced in 1956 as a non-flammable, non-explosive, halogenated volatile anesthetic that is usually mixed with air or oxygen.
- The presence of the carbon-halogen bonds contributes to its non-flammability, volatility, and high lipid solubility (Blood/Gas partition coefficient = 2.3).
- It activates GABAA and glycine receptors. It also acts as an NMDA receptor antagonist and inhibits nACh and voltage-gated sodium channels.
Structure Activity Relationship:
- The SAR studies conducted independently by Meyer and Overton in the 1880s showed a distinct positive correlation between anesthetic potency and solubility.
- The potency of alkanes, cycloalkanes, and aromatic hydrocarbons increases in direct. proportion to the number of carbon atoms in the structure upto a cutoff point 7. Within the n-alkane series, the cutoff number is 10, with n-decane showing minimal anesthetic potency.
Metabolism:
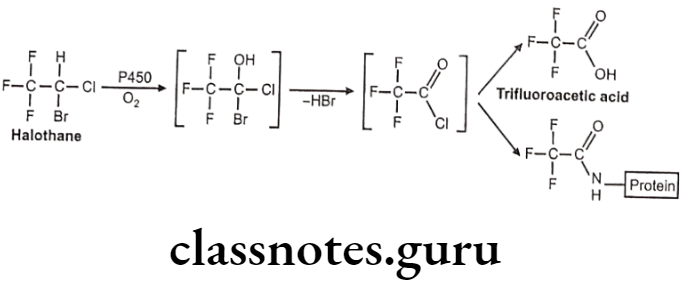
Uses:
- Halothane can be used to start or maintain anesthesia.
- One of its benefits is that it does not increase the production of saliva which can be particularly useful in those who are difficult to intubate.
General Structure-Activity Relationship of Alkanol Series:
- Similar increase in potency with increase in carbon length was seen in the n-alkanol series.
- In association, the n-alkanol with a given a number of carbons is more potent than the n-alkane with same chain length.
Effect of Halogenation on Ether:
- Decrease the flammability of the ether, enhances their stability and increases their potency.
- Higher atomic mass halogens increase potency more compared to lower atomic mass halogens. e.g. Replacing the fluorine (F) in desflurane with chorine (CI) to form isoflurane increases the potency more than four-fold.
- Replacing the chlorine (CI) with bromine (Br) increases the potency three-fold i.e. isoflurane.
- Unfortunately, halogenation also increases the toxicity.
- Halogenated methyl ethyl ether was found to be more stable and potent than halogenated diethyl ether. i.e. enflurane and isoflurane.
- For n-alkane series, fully saturating the alkane with fluorine (F) increases the potency. Number of carbon atoms was 2 to 4 which show highest potency. If it is more than 5, then the activity is diminished..
- The stereoisomer of isoflurane (+) and (-) has been isolated and tested for anesthetic potency. The (+) isomer was found to be 53% more potent than (-) isomer.
- The addition of double and triple bonds to anesthetics molecule decreases their potency.
Methoxyflurane:
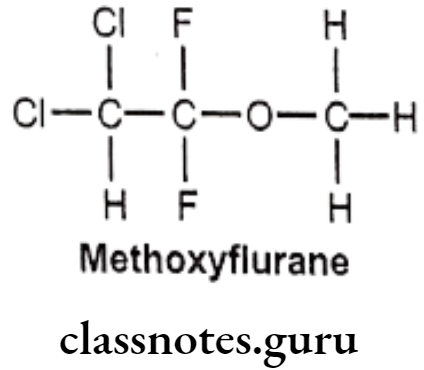
- Methoxyflurane is chemically 2,2-dichloro-1,1-difluoro-1-methoxyethane.
- It acts as a positive allosteric modulator of the GABAA receptor, it also acts as an NMDA receptor antagonist.
- It should be administered with nitrous oxide to achieve a relatively light level of anesthesia, and a neuromuscular blocking agent given concurrently to obtain the desired degree of muscular relaxation.
Metabolism:
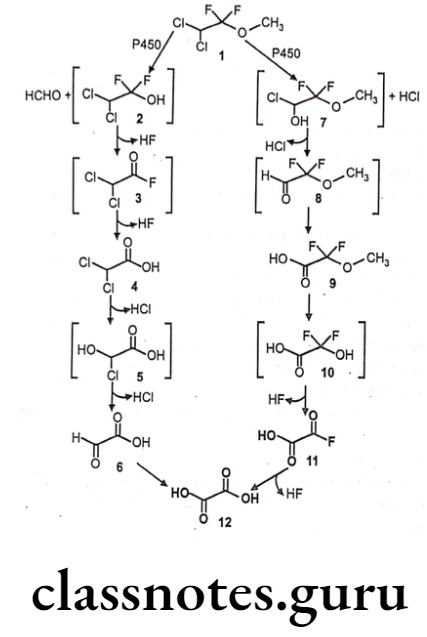
Uses:
- Methoxyflurane provides rapid short-term analgesia using a portable inhaler device.
- Currently, methoxyflurane is rarely used for surgical, obstetric, or dental anesthesia.
Enflurane:
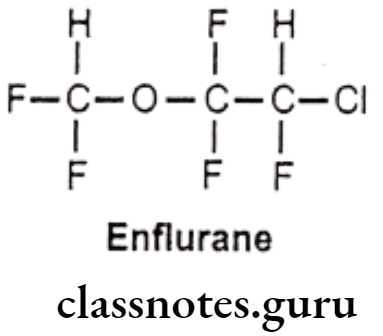
- Enflurane is chemically 2-chloro-1-(difluoromethoxy)-1,1,2-trifluoroethane.
- It acts as a positive allosteric modulator of the GABAA, glycine, and 5-HT3 receptors.
- It is a fluorinated ether and very potent general anesthetic agent.
- It is more stable inhalation anesthetic which provides rapid adjustments of anesthesia depth with little change in pulse or respiratory rate.
Metabolism:
- Enflurane is metabolized via CYP2E1 to form a fluoride ion and difluoromethoxy difluoro- acetic acid metabolites.
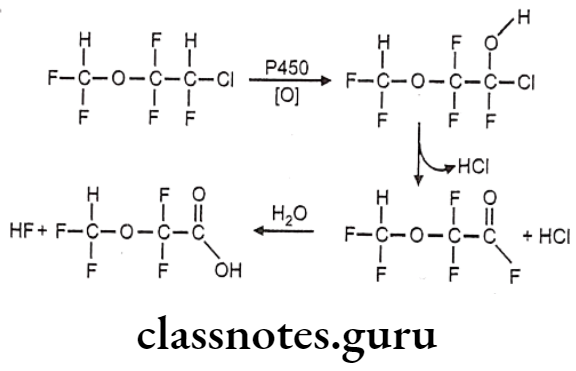
Uses:
- Enflurane may be used for induction and maintenance of general anesthesia.
- It can also be used to provide analgesia for vaginal delivery.
Isoflurane:
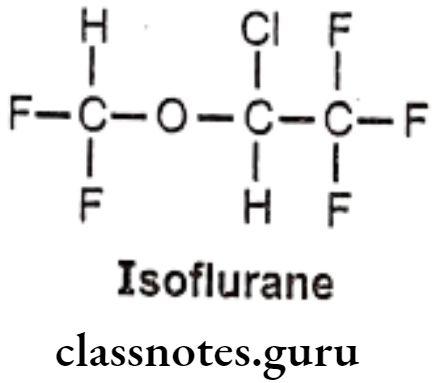
- Isoflurane is an isomer of enflurane.
- It is chemically 2-chloro-2-(difluoromethoxy)-1,1,1-trifluoroethane.
- It is a fluorinated ether with general anesthetic and muscle relaxant activities.
- It reduces pain sensitivity (analgesia) and relaxes muscles.
- It likely binds to GABA, glutamate and glycine receptors, but has different effects on each receptor.
- It acts as a positive allosteric modulator of the GABAA receptors.
- It inhibits receptor activity in the NMDA glutamate receptor subtypes.
- It inhibits conduction in activated potassium channels.
- It also affects intracellular molecules and NADH dehydrogenase.
Metabolism:
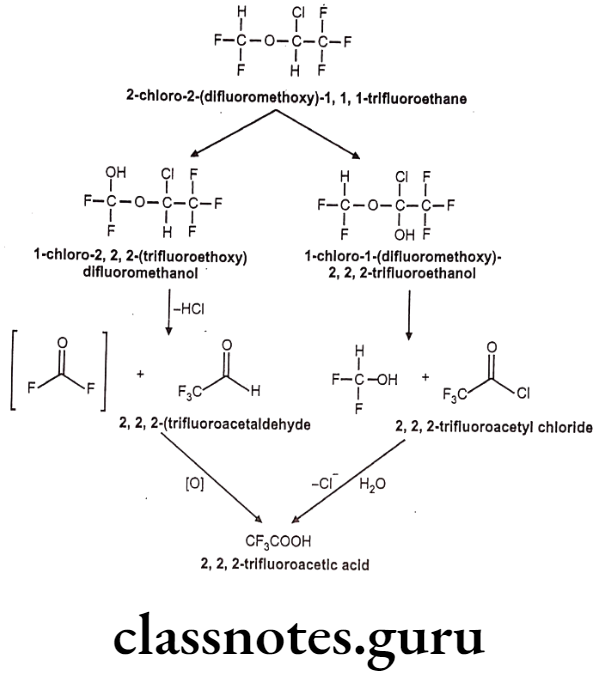
Uses:
- Isoflurane is a general anesthetic.
- It can be used to start or maintain anesthesia.
- Often another medication is used to start anesthesia due to airway irritation with isoflurane.
Sevoflurane:
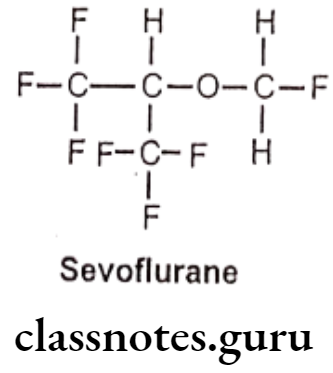
- Sevoflurane is chemically, 1,1,1,3,3,3-hexafluoro-2-(fluoromethoxy)propane.
- It is a fluorinated isopropyl ether with general anesthetic property.
- It acts as a positive allosteric modulator of the GABAA receptor, it also acts as an NMDA receptor antagonist.
Metabolism:
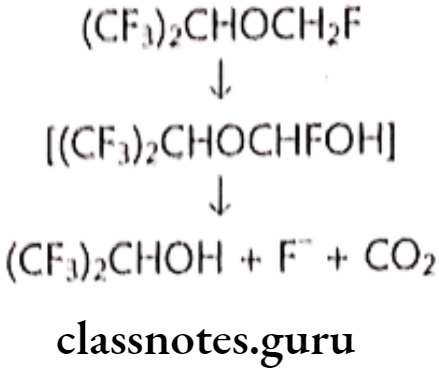
Uses:
- Sevoflurane is one of the most commonly used volatile anesthetic agents, particularly for outpatient anesthesia, across all ages, as well as in veterinary medicine.
Desflurane:
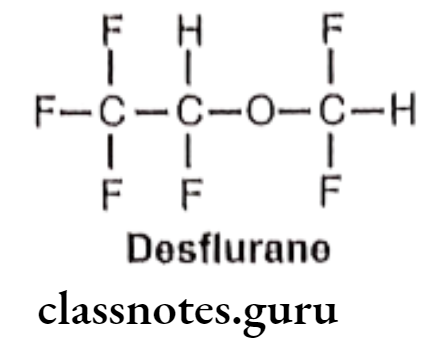
- Desflurane is chemically, 2-(difluoromethoxy)-1,1,1,2-tetrafluoroethane.
- It is a highly fluorinated methyl ethyl ether used for maintenance of general anesthesia.
- It acts on the lipid matrix of the neuronal membrane, resulting in disruption of neuronal transmission in the brain.
- It may also enhance the synaptic activity of the inhibitory neurotransmitter gamma- aminobutyric acid (GABA).
- It may activate GABA channels and hyperpolarize cell membranes.
- It also may inhibit certain calcium channels and therefore prevent release of neurotransmitters and inhibit glutamate channels.
Metabolism:
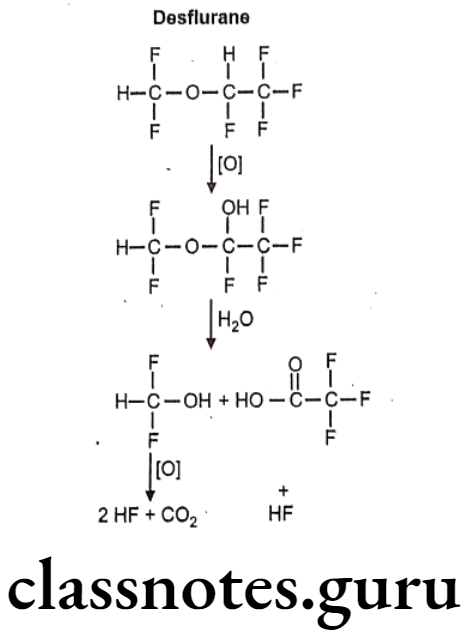
Uses:
- Drugs Acting on Central Nervous System (II)
- Desflurane is a non-flammable liquid general anesthetic administered via vaporizer. • It is indicated as an inhalation agent for induction of anesthesia for inpatient and outpatient surgery in adults.
Ultra Short-Acting Barbiturates
Thiopental sodium:
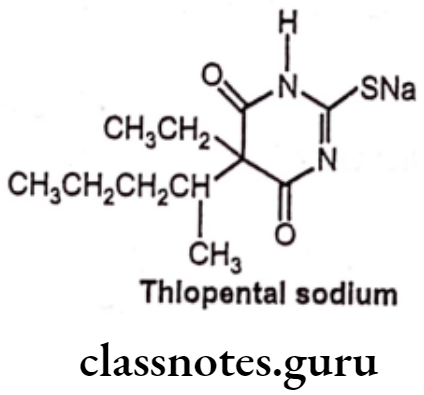
- Thiopental sodium is chemically, 5-ethyl-5(1-methylbutyl)-2-thiobarbiturate.
- Thiopental binds to the chloride ionophore site of the gamma-aminobutyric acid GABA/chloride ionophore receptor complex, thereby enhancing the inhibitory actions of GABAA in the brain which leads to synaptic inhibition, decreased neuronal excitability and induction of anesthesia.
- It also decreases glutamate responses.
Metabolism:
- Thiopental sodium is a barbiturate that is administered intravenously for the induction of general anesthesia of short duration.
- It helps patients to relax before receiving general anesthesia with an inhaled medication.
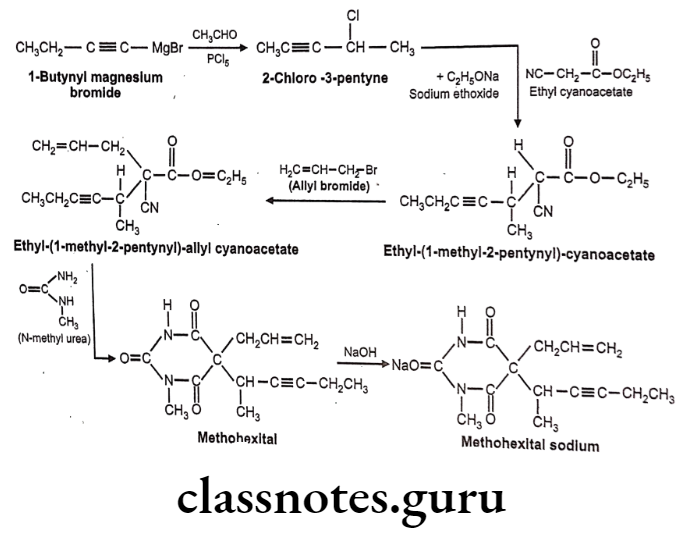
Methohexital sodium:
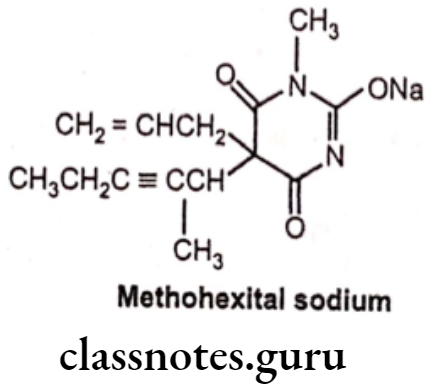
- Methohexital sodium is chemically sodium, 5-(hex-3-yn-2-yl)-1-methyl-2,6-dioxo- 5-(prop-2-en-1-yl)-1,2,5,6-tetrahydropyrimidin-4-olate.
- It binds to a distinct site which is associated with CI ionophores at GABAA receptors. This increases the length of time for which the CI ionopores are open, thus causing an inhibitory effect.
Metabolism:
- Metabolism of methohexital is primarily hepatic via demethylation and oxidation. Side-chain oxidation is the primary means of metabolism involved in the termination of the drug’s biological activity.
Uses:
- Methohexital sodium is an intravenous anesthetic with a short duration of action that may be used for induction of anesthesia.
- It has been commonly used to induce deep sedation or general anesthesia for surgery and dental procedures.
Thiamylal Sodium:
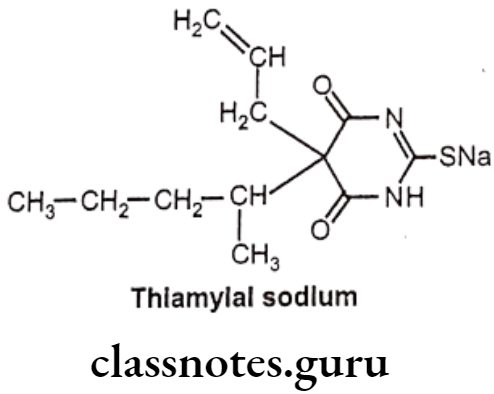
- Thiamylal sodium is chemically sodium – 4,6-dioxo-5-pentan-2-yl-5-prop-2-enyl-1H- pyrimidine-2-thiolate.
- It binds at a distinct binding site associated with a CI ionopore at the GABAA receptor, increasing the duration of time for which the CI ionopore is open. The post-synaptic inhibitory effect of GABA in the thalamus is, therefore, prolonged.
Uses:
- Thiamylal sodium is a barbiturate that is administered intravenously for the production of complete anesthesia of short duration.
- It has sedative, anticonvulsant, and hypnotic effects, and is used as a strong but short acting sedative.
- It is still in current use, primarily for induction in surgical anesthesia or as an anticonvulsant.
Dissociative Anesthetics
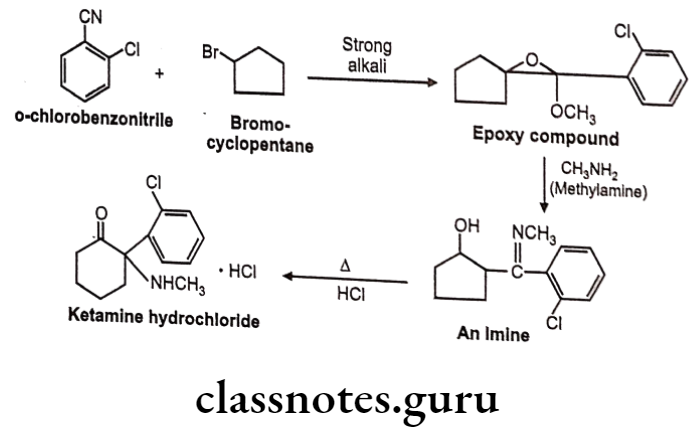
Ketamine Hydrochloride:
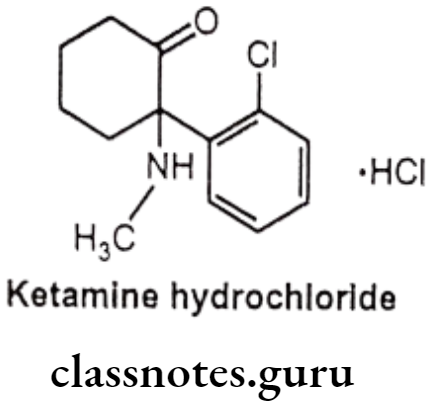
- Ketamine hydrochloride is chemically 2-(2-chlorophenyl)-2-(methylamino) cyclohexan-1-one;hydrochloride.
- It interacts with N-methyl-D-aspartate (NMDA) receptors, opioid receptors (μ and σ), monoaminergic receptors, muscarinic receptors and voltage sensitive Ca+ ion channels and thereby reducing pain perception, inducing sedation, and producing dissociative anesthesia.
- Unlike other general anesthetic agents, ketamine does not interact with GABA receptors.
Metabolism:
- Ketamine presents a mainly hepatic metabolism and its major metabolite is nor-ketamine. The biotransformation of ketamine corresponds to N-dealkylation, hydroxylation of the cyclohexone ring, conjugation to glucuronic acid and dehydration of the hydroxylated metabolites for the formation of cyclohexene derivatives.
Uses:
- Ketamine hydrochloride is a cyclohexanone derivative used for induction of anesthesia.
- Anesthesia in children, as the sole anesthetic for minor procedures or as an induction agent followed by muscle relaxant and tracheal intubation.
- It can be given to asthmatics or people with chronic obstructive airway disease.
- It acts as a sedative for physically painful procedures in emergency departments.
Synthesis
Halothane
Methohexital Sodium
Ketamine Hydrochloride
Multiple Choice Questions:
Question 1. The state of “general anesthesia” usually includes:
- Analgesia
- Loss of consciousness, inhibition of sensory and autonomic reflexes
- Amnesia
- All of the above
Answer. 4. All of the above
Question 2. The IUPAC name of Halothane is
- 2-bromo-2-chloro-1,1,1-trifluoroethane
- 2-bromo-2-chloro-1,1, -difluoroethane
- 2-bromo-1,1,1-thrifluoroethane
- None of above
Answer. 1. 2-bromo-2-chloro-1,1,1-trifluoroethane
Question 3. Inhaled anesthetics and intravenous agents having general anesthetic properties:
- directly activate GABAA receptors
- facilitate GABA action but have no direct action on GABAA receptors
- reduce the excitatory glutamatergic neurotransmission
- increase the duration of opening of nicotine-activated potassium channels
Answer. 1. directly activate GABAA receptors
Question 4. Indicate the anesthetic, which is an inhibitor of NMDA glutamate receptors:
- Thiopental
- Halothane
- Ketamine
- Sevoflurane
Answer. 3. Ketamine
Question 5. Halothane contains the haloatoms
- Bromine, chlorine and fluorine
- Bromine, chlorine and iodine
- Bromine and iodine
- Chlorine and fluorine
Answer. 1. Bromine, chlorine and fluorine
Question 6. An ideal anesthetic drug would:
- induce anesthesia smoothly and rapidly and secure rapid recovery
- possess a wide margin of safety
- be devoid of adverse effects
- All of the above
Answer. 4. All of the above
Question 7. Which of the following general anesthetics belongs to inhalants?
- Thiopental
- Desfluran
- Ketamine
- Propofol
Answer. 2. Desfluran
Question 8. Which of the following inhalants is a gas anesthetic?
- Halothane
- Isoflurane
- Nitrous oxide
- Desflurane
Answer. 3. Nitrous oxide
Question 9. Indicate the intravenous anesthetic, which is an ultra-short-acting barbiturate:
- Fentanyl
- Thiopental
- Midazolam
- Ketamine
Answer. 2. Thiopental
Narcotic And Non-Narcotic Analgesics
Introduction
Pain is the most common complaint for which patients seek treatment. Pain has been classified into various types such as physiological (e.g. touching a hot object or getting a cut), inflammatory (e.g. infection and tissue injury), and neuropathic (e.g. injury to the PNS or CNS).
Agents that decrease pain are referred to as analgesics, or analgetics or pain-killing drugs. As these drugs act as pain relieving agents, they are also called antinociceptives (reduced sensitivity of pain).
There are number of classes of drugs which relieve pain. Mainly these drugs are categorized into two classes, (a) morphine and related compounds and (b) antipyretics and anti-inflammatory analgesics.
Non-Steroidal Anti-Inflammatory Agents (NSAID), have primarily a peripheral site of action and are useful to relive mild to moderate pain without loss of consciousness. These agents often have an anti-inflammatory effect associated with their pain killing action.
Historically, opioid analgesics have been called narcotic analgesics. Narcotic analgesics are selective CNS depressant that cause sleep or loss of consciousness (narcosis) in conjugation with their analgesic effect. These are extremely potent analgesics and are effective for the relief of severe pain. Not all opioid analgesics cause necrosis.
The side effects caused by opioid analgesics are respiratory depression, nausea and drowsiness. Long term administration may lead to tolerance, psychological and/or physical dependence called as addiction.
Because of addiction properties, opioid class has been the most problematic for use in the proper management of pain. The term opiophobia was coined to describe the reluctance of physicians to prescribe opioid drugs in adequate amounts or for long enough periods.
Opioid Receptors:
The neuronal located proteins to which opioid agent binds and initiate biological response are called as opioid receptors. There are three major types of receptors classified by the order in which they are cloned.
- Delta (8) receptors are PO1 receptors.
- Kappa (K) receptors are PO2 receptors.
- Mu (u) receptors are PO3 receptors.
All of these receptors are located in human brain or spinal cord tissues and each has a role in the mediation of pain. These receptors have subtypes that provide varying degrees of analgesia, euphoria or dysphoria, central nervous system depression, and perhaps, the potential for tolerance.
Orphan opioid receptor (fourth receptor) has been identified and cloned (OP4) based on homology with cDNA sequence of the known (μ, 8 and K) opioid receptors. Despite the homology in cDNA sequence with known opioid receptors, this new receptor did not bind the classical peptide or non-peptide agonists or antagonists with high affinity.
Mechanism of Action of Narcotic Analgesics
The signal transition mechanism for (u, 8 and k) opioid receptors is through G-proteins. Activation of opioid receptors is linked through the G-protein to an inhibition of adenylate cyclase activity. This results into decrease in cAMP production, efflux of K* and closure of voltage gated Ca** channel leads to hyperpolarization of the nerve cell and strong inhibition of nerve firing.
Classification Of Narcotic Analgesics
- Natural alkaloids: Morphine, Codeine.
- Semi-synthetic analogues: Hydromorphone, oxymorphone, oxycodone.
- Synthetic agents: Meperidine, Methadone, Phenazocine, Pentazocine, Fentanyl Dextropropoxyphene, Nalorphine, Naloxone, Naltrexone.
Morphine and Related Drugs:
The prototypic narcotic analgesic is (-)-morphine, the principal alkaloid obtained from juice or latex from the unripe seeds of the poppy plant, (Papaver somniferum). The first alkaloid to be isolated was morphine (10% in opium), which then underwent structural modifications to produce a number of derivatives of morphine. It also contains codeine. (0.5%), thebaine (0.2%), papaverine (1%) and noscapine (6%).
Morphine Analogues
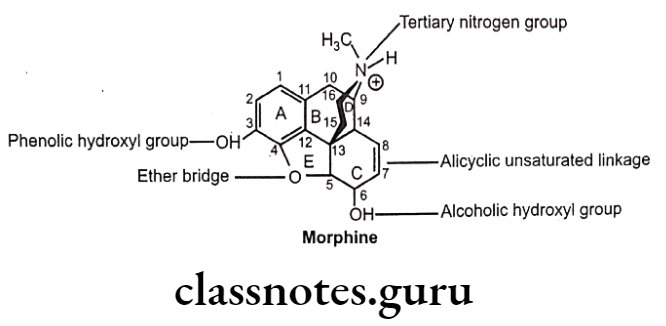
Structure-Activity Relationship
- Morphine is a prototype opioid.
- It is selective for μ opioid receptor.
- Structure of morphine composed of five rings (fused) i.e. a benzene ring (A), two partially unsaturated cyclohexane rings (B and C), a piperidine ring (D) and a dihydrofuran ring (E).
- Morphine molecule has 5 chiral centers with absolute stereochemistry, 5(R), 6(S), 9(R), 13(S) and 14(R).
- Naturally occurring morphine is levo (-) rotatory.
- Dextro (+) morphine has been synthesized and it is devoid of analgesic and other opioid activity.
- Any major change will cause changes in the affinity and intrinsic activity of the new compound at each opioid receptor type.
- Thus opioid receptor selectivity profile of new compound may be different than the structure from which it was made (e.g. from μ 8 or K).
- It may also have different physiological properties such as solubility, partition coefficient, pKa etc. results in different pharmacokinetic characteristics and can affect its in-vitro activity profile.
Ring-A:
- Ring ‘A’ and basic ‘N’ exist in protonated (ionized) form, required for opioid analgesic activity.
- Aromatic ring ‘A’ and cationic ‘N’ may be connected by ethyl linkage (i.e. 9, 10 position of B-ring) or propyl linkage (either edge of the piperidine ring that forms the D-ring).
- ‘A’ ring and basic ‘N’ are necessary components for every potent μ- agonist known. (iv) These two alone are not sufficient for μ-activity, however an additional pharmacophores are required such as,
- Rigid structure (i.e. fused ring A, B, and D)
- 3-OH and tertiary ‘N’ either greatly enhance or are essential for activity.
N-atom:
- Tertiary ‘N’ group has good opioid activity.
- Size of ‘N’ substitutions indicate agonist or antagonist properties.
- N-CH3 substitution has good agonist activity.
- Increase in ‘N’ substitution to 3-5 carbon (unsaturated or smaller carbocyclic ring), antagonist at some or all receptor types.
- Longer substitution on ‘N’ returns agonist properties to the opioid.
- N-phenyl ethyl substitution opioid is usually 10-fold more potent as a μ-agonist than the N-CH3 analogue.
- N-CH2-CH=CH2 substitution leads to μ-antagonist activity (Naloxane).
3-phenolic hydroxyl group:
- Substitution of 3-H instead of 3-OH decreases the activity 10 times.
- Substitution of 3-CH3 in place of 3-OH decreases analgesic activity, the compound so formed possesses anti-tussive activity (i.e. Codeine).
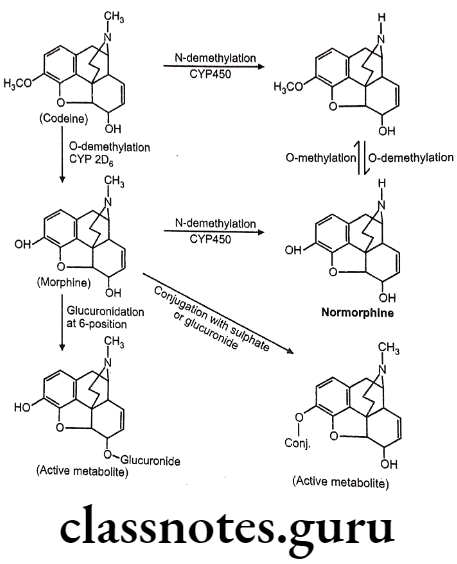
(Codeine is weak μ-agonist and it undergoes slow o-demethylation to morphine).
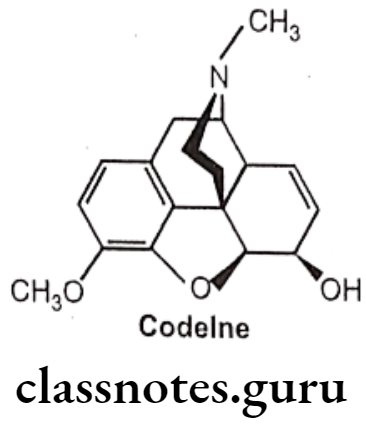
- Substitution of ester (CH3-CO-) at position 3 decreases the activity.
- Diacetyl group at 3 and 6 position leads to formation of Heroin, which is synthesized in 1874 and marketed in 1898 by Friedrich Bayer Co. in Germanay.
- Heroin is preferable to morphine as it does not disturb digestion or produce habit readily.
- It has low affinity for u-receptors.
- It has high lipophilicity compared to morphine and easily cross blood-brain barrier.
- In body (including the brain), serum and tissue esterase hydrolyse 3-acetyl group to produce 6-acetyl morphine leads to increase μ-receptor activity in excess of morphine.
- Administration of heroin by IV route rapidly converted to a potent μ-agonist provide a ‘euphoric rush’ – popular drug of abuse.
- Repeated use of heroin develops tolerance, physical dependence and acquisition of the drug habit.
- Self administration of heroin with unclean or shared hypodermic needle, results in transmission of HIV, hepatitis or other infectious disease.
C-ring:
- 6-keto decreases the activity in pure morphine only.
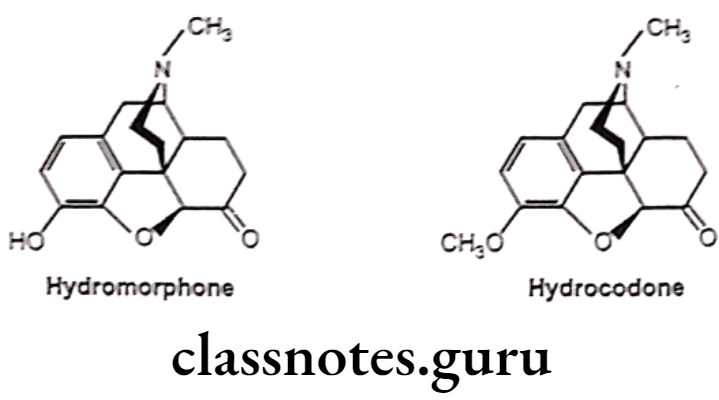
- 7,8-dihydro (no double bond between 7 and 8 carbon atom) with 6-keto derivative of morphine increases activity by 8-10 times than morphine (i.e Hydromorphone).
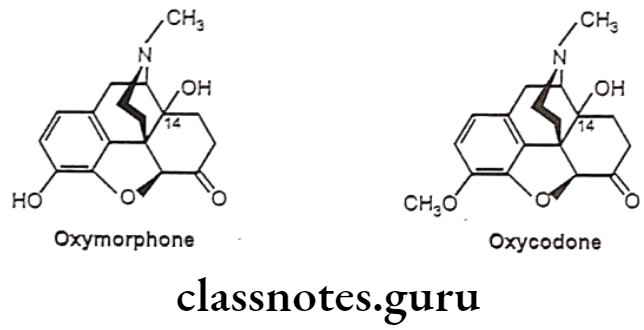
- Substitution of 3-OCH; derivative of hydromorphone (hydrocodone) is more active than codeine.
- Substitution of 6-H decreases the activity.
14a-hydroxy-6-keto derivatives:
- Substitution of B-OH at 14 position increases the analgesic activity (oxymorphone).
- Substitution of 3-OH and N-CH3 derivative is 10 times more potent than morphine.
- Substitution of 3-OCH3 derivative of oxymorphone (Oxycodone) is as potent as morphine when given parenterally, but oral dose is better than parenteral compared to morphine.
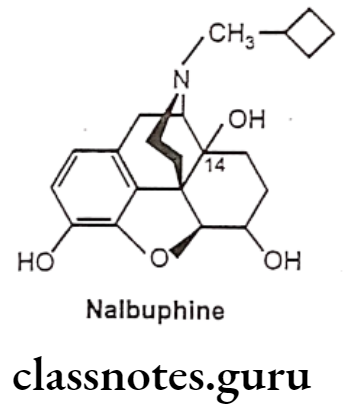
- Substitution of N-cyclobutyl methyl and reduction of 6-keto to 6a-OH of oxymorphone (Nalbuphine) acts through K-receptor and possess approximately half analgesic potency of morphine. Nalbuphine acts as a μ-receptor antagonist.
- Replacement of the potent narcotic agonist oxymorphone’s N-methyl group with an allyl group (-CH2-CH=CH2) (Naloxone) or methyl cyclopropyl group (Naltrexone) acts as pure opioid antagonist. Both the drugs act as antagonist of all types of opioid receptor.
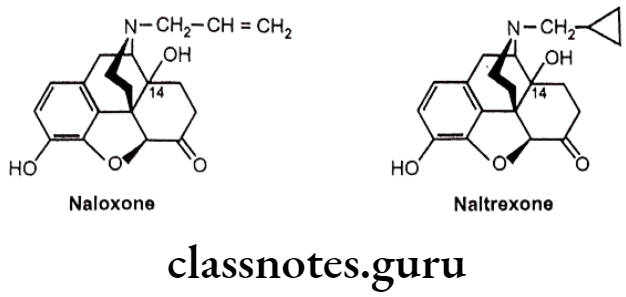
- Substitution of -OH group at C14 and keto group at C6 is necessary for pure antagonist activity.
3,4-Epoxide Bridge (Morphinans):
Removal of 3,4-epoxide bridge (4-5 ether bridge) in the morphine structure results in compounds called as morphinans.
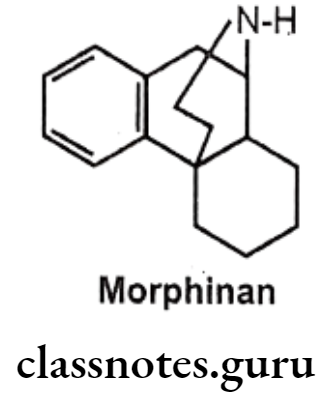
- Morphinan can be prepared by synthetic route only, as it is very difficult to remove it from morphine analogue.
- The synthetic procedure yields compounds as racemic mixtures in which only the levo (-) isomers act as opioid analgesic, whereas dextro (+) isomers act as anti-tussive.
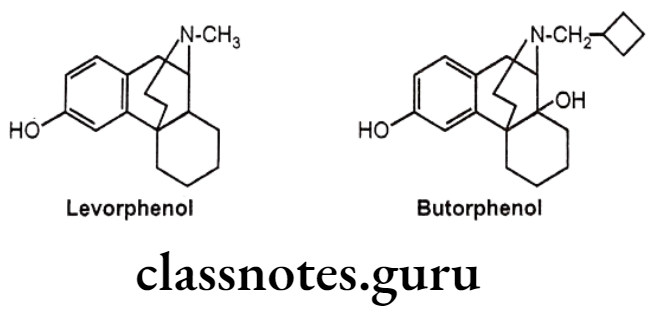
- Morphinan derivative,
- Levorphenol possesses 8 times more potent analgesic activity as compared with morphine due to increased μ-receptor affinity and increased lipophilicity.
- Butorphenol have mixed agonist/antagonists activity. It is a u-receptor antagonist and K-receptor agonist.
Benzomorphans:
- Benzomorphans are synthetic compounds that lack both the epoxide E ring and C ring of morphine, having only A, B and D rings, retain opioid activity.
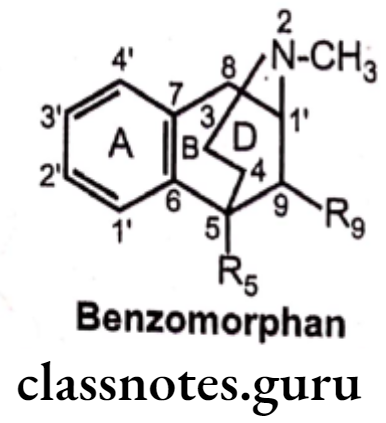
- They are named chemically as benzomorphan or use a different nomenclature system as 2, 6-methano-3-benzazocine.
- Analgesic activity increases in order of substitution of OH>H>NH2, NO2, F and Cl at 2′ position.
- Trimethyl substitution at N=CH3, R5-CH3 and Rg-CH3 possesses 3 times more potent analgesic activity than dimethyl Rs-H and R9 CH3.
- Substitution of R-CH3 increases the analgesic activity.
- Substitution of R-OH having the same activity as substitution of -OH at 14th position of morphine leads to decease in analgesic activity.
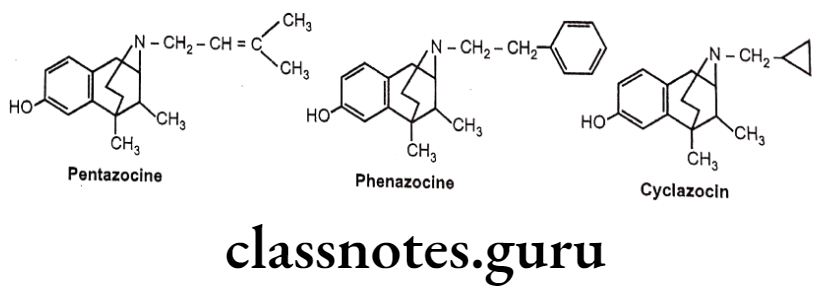
- Pentazocine is a weak μ-receptor antagonist, whereas it is a k-receptor agonist and thus produces analgesia.
- Phenazocine (N-Phenylethyl substituted benzomorphan) is 10 times more potent than morphine as a μ- agonist.
- Cyclazocine (N- Cyclopropyl methyl substituted benzomorphan) is a narcotic antagonist. It possesses considerable hallucinogenic properties.
4-Phenylpiperidines:
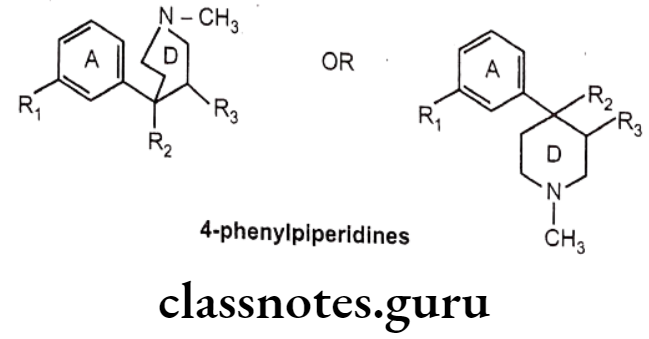
- 4-phenylpiperidines possess only A and D ring analogues of morphine.
- The first agent among this class, meperidine, was synthesized in 1937 by Eislab, who was attempting to prepare antispasmodic agents, but serendipitously observed the opioid activity.
- Meperidine is proved to be a typical μ-agonist, with approximately 1/4th potency of morphine. It have very short duration of action because of its esterases hydrolysis to a zwitter ionic metabolite.
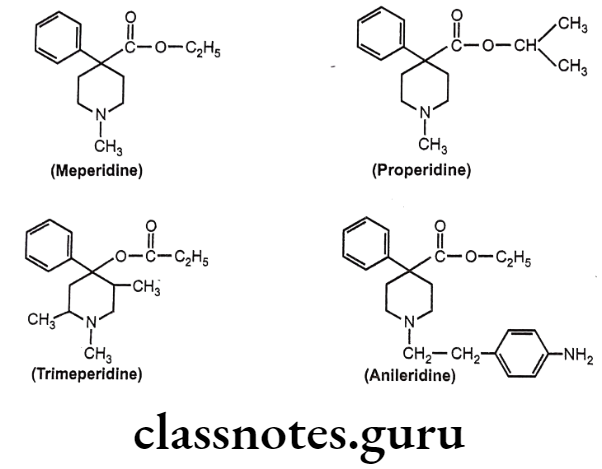
- Replacement of ethyl group of meperidine with isopropyl group (properidine), have 15 times more potent activity than meperidine.
- Reversed esters of meperidine have greater potency. 1,2,5- trimeperidine (propionoxy compound) is 7.5 times more potent than meperidine.
- Replacement of N-CH3 group of meperidine with N-p-amino phenyl ethyl group (Anileridine) is 4 times more potent than meperidine.
Anilinopiperidines:
- Structural modification of the 4-phenylpiperidines results into discovery of new compounds, 4-anilidopiperidine or the fentanyl.
- In fentanyl, phenyl and acyl groups are separated from the ring by nitrogen.
- Fentanyl and its derivatives are μ-receptor agonists.
- They are powerful analgesic (50 times stronger than morphine) with minimum side effects.
- Its short duration of action makes it well suited for use in anesthesia. pa
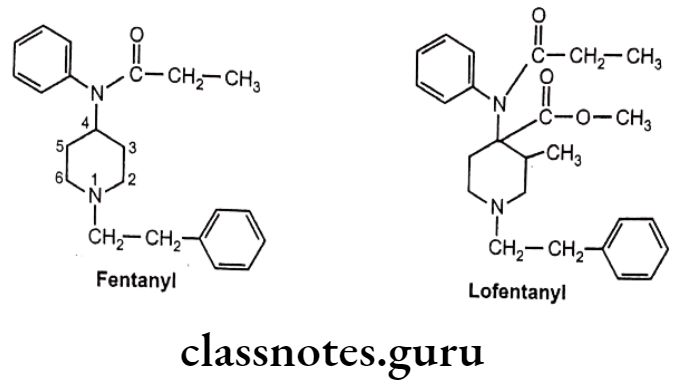
- Substitution of -CH3 at 3-postion of piperidine ring with addition of a small oxygen containing group at the 4-position of the piperidine ring, leads to the formation of analogues with extreme potent analgesic activity. (i.e. Lofentanyl – 8,400 times potent than morphine).
- Alfentanyl, sufentanyl and remifentanyl are the other examples of fentanyl derivatives, which are having highly safety margin than other μ-agonists.
Diphenylheptanone:
- German scientists synthesized another series of open-chain compounds as potential antispasmodics.
- When the nitrogen ring of morphine is opened, the analgesic activity is virtually abolished.
- Further modifications by scientists made the compound to possess both analgesic and spasmolytic activity.
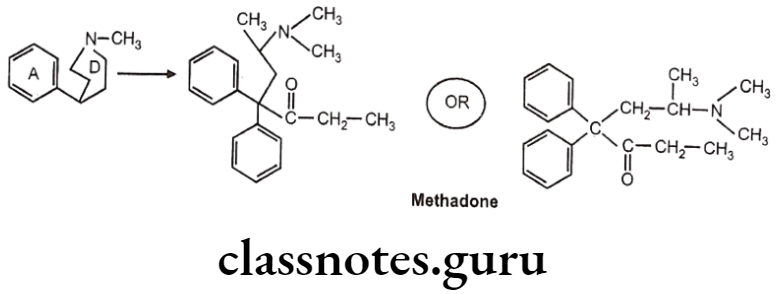
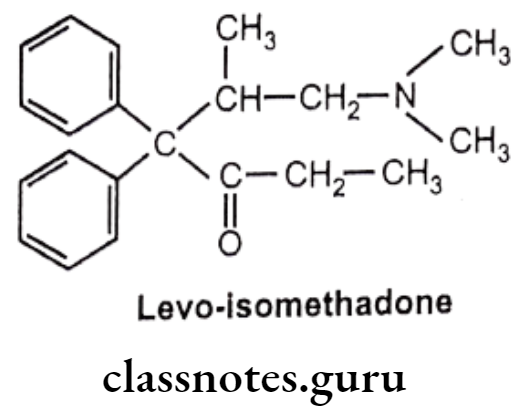
- Methadone was the first analgesic agent..
- Levo isomer of methadone and levo-isomethadone are twice active as their recimic mixture.
- Reduction of keto group leads to decrease in activity.
- Removal of any phenyl group leads to decrease in activity.
- Replacement of dimethylamino group with pyrrolidyl group gives 3/4th activity as methadone.
Metabolism of Methadone:
Metabolism of Opioids (In liver):
Morphine sulphate:
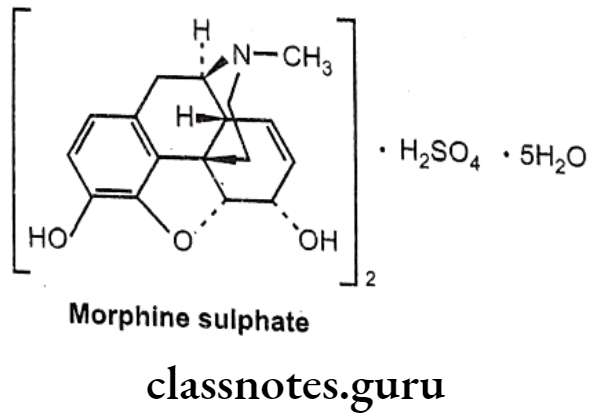
- The prototypic narcotic analgesic is (-)-morphine, the principal alkaloid obtained from the Opium poppy (Papaver somniferum).
- Morphine was isolated as a pure alkaloid by a German Pharmacist, Serturner in 1803. Morphine sulphate is chemically, (4R,4aR,75,7aR,12bS)-3-methyl-2,4,4,7,7,13- hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinoline-7,9-diol;sulphuric acid.
- It exerts the major effects by interacting with opioids receptors (μ, 8 and K) in the CNS.
- It activates 7 TM GPCRs located presynaptically and postsynaptically along pain transmission pathways.
Uses:
- Morphine is used as narcotic analgesic.
- It is used as pre-anesthetic medication.
- It can also be used for the treatment of diarrhoea.
- It produces sleep and sedation.
Codeine:
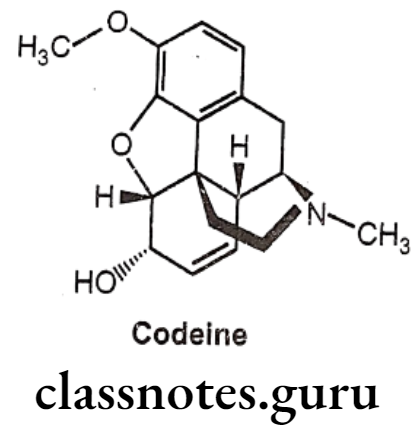
- Codeine is an alkaloid that occurs in opium, but the amount present is usually too small to be of commercial importance.
- Conversion of the 3-OH to a 3-OCH3, yields codeine, reduces activity to 15% of morphine.
- It is chemically, (4R,4aR,75,7aR,12bS)-9-methoxy-3-methyl-2,4,4a,7,7a,13-hexahydro-
- 1H-4,12-methanobenzofuro[3,2-e]isoquinoline-7-ol.
- It is available as a sulphate and phosphate salt and also as the free base and as tablets, elixir and solution for injection.
- The 3-methoxy group protects the 3-position from glucuronide as occurs with morphine.
Uses:
- Codeine is used as an analgesic, antitussive and cough suppressor.
- It also possesses antidiarrheal and antihypertensive activity.
- It has hypnotic and sedative property and can be used as antianxiety drug.
Meperidine Hydrochloride:
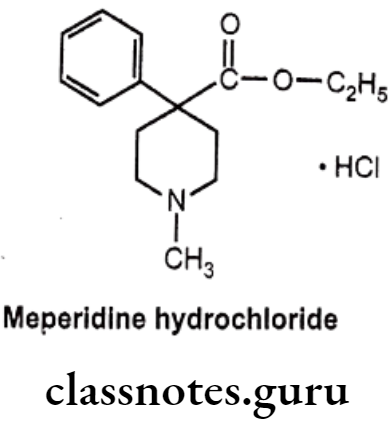
- Meperidine hydrochloride is chemically ethyl-1-methyl-4-phenylpiperidine-4- carboxylate; hydro-chloride.
- It has very short duration of action and largely metabolized in the liver with only a small quantum of it~ 5% gets excreted unchanged.
- Importantly, the ‘esterases’ predominantly cause cessation of the ester linkage (as ethyl ester at para-position) to leave as residue the inactive-carboxylate analogue.
- It also undergoes N-demethylation to yield the corresponding product known as ‘normeperidine’ – a metabolite which gets accumulated after a prolonged medication with meperidine.
Uses:
- Meperidine is a narcotic analgesic that can be used for the relief of most types of moderate to severe pain, including postoperative pain and the pain of labour.
- It can also use as a premedication before anesthesia.
Anileridine Hydrochloride:
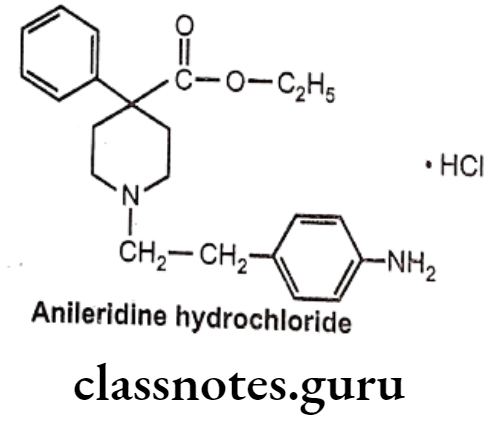
- Anileridine is chemically, ethyl 1-[2-(4-aminophenyl)ethyl]-4-phenylpiperidine-4- carboxylate; hydrochloride.
- It is a synthetic analgesic drug and is a member of the piperidine class of analgesic agents.
Uses:
- It differs from pethidine (meperidine) in that the n-methyl group of meperidine is. replaced by an N-aminophenethyl group, which increases its analgesic activity.
Diphenoxylate Hydrochloride:
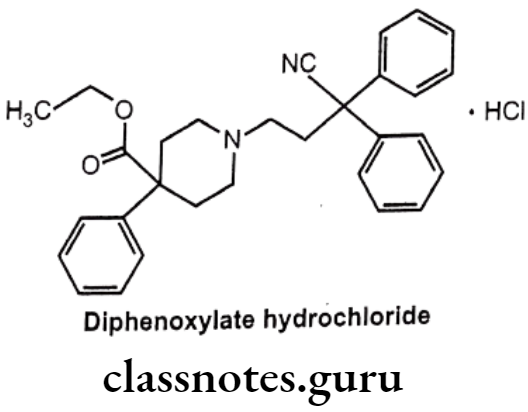
- Diphenoxylate is chemically, ethyl-1-(3-cyano-3, 3-diphenylpropyl)-4-phenyl- isonipecotate.
- Diphenoxylate is a narcotic antidiarrheal drug related chemically to loperamide.
- It reduces bowel contractions and consequently the frequency and fluidity of bowel movements.
- Although diphenoxylate is chemically related to narcotics, it does not have pain relieving (analgesic) actions like most other narcotics.
- In higher doses, like other narcotics, diphenoxylate can cause euphoria (elevation of mood) and physical dependence.
Uses:
- It is a synthetic analogue of pethidine (meperidine) with some analgesic activity, but is mostly used in the treatment of diarrhoea associated with gastroenteritis, irritable bowel, acute infections, hyper motility, ulcerative colitis and sometimes even food poisoning.
- It prevents hyper gastrointestinal propulsion by reducing intestinal motility.
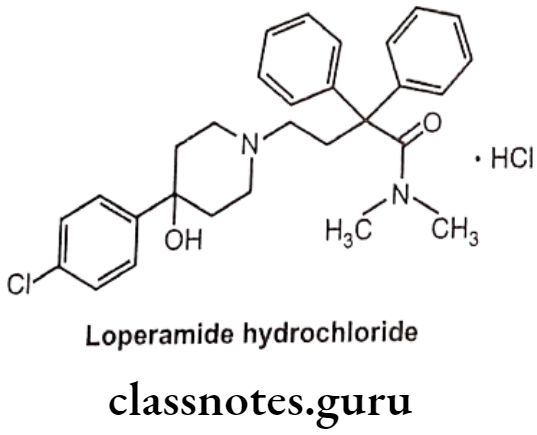
Loperamide Hydrochloride:
- Loperamide hydrochloride is loperamide, a synthetic, piperidine derivative.
- It is chemically, 4-(4-chlorophenyl)-4-hydroxy-N,N-dimethyl-a,a-diphenyl-1-piperidine butyramide hydrochloride.
- It acts on the μ-receptors in the intestinal mucosa.
- It slows intestinal motility by acting on the nerve endings and/or intramural Ganglia embedded in the intestinal wall.
- The prolonged retention of the feces in the intestine results in reducing the volume of the stools, increasing viscosity, and decreasing fluid and electrolyte loss.
Uses:
- Loperamide hydrochloride is used as opioid agonist.
- It is used in symptomatic relief of acute non-specific diarrhoea and of chronic diarrhoea associated with inflammatory bowel disease.
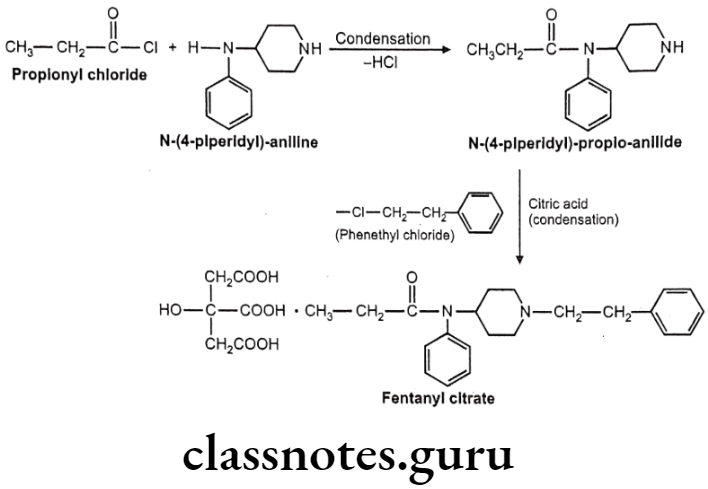
Fentanyl citrate:
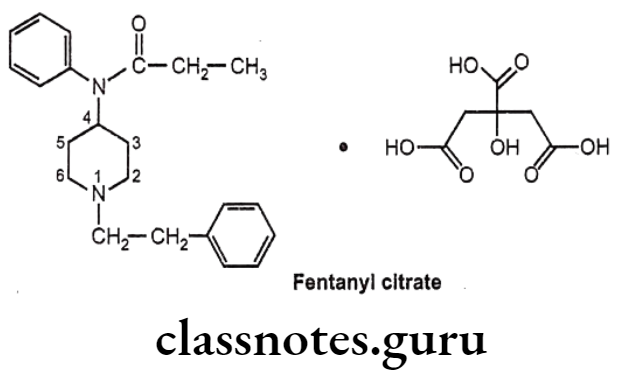
- Fentanyl citrate is chemically N-phenyl-N-[1-(2-phenylethyl)piperidin-4-yl] propanamide-N-(1-phenylethyl-4-piperidinyl) propionanilide, citrate.
- It is a very potent synthetic opiate, which can be used, as an analgesic.
- It is structurally related to phenylpiperidines (e.g. meperidine) and produces strong analgesia, similar to morphine.
- It possesses an inherent rapid onset and short duration of action.
Uses:
- Fentanyl is 80 times more potent than morphine as analgesic primarily employed as an analgesic for the arrest of pain after all types of surgical procedures.
- It is used as an aid for induction and maintenance of inhalation anesthesia.
- It may be employed also as an adjuvant to all such drugs mostly used for regional and general anesthesia.
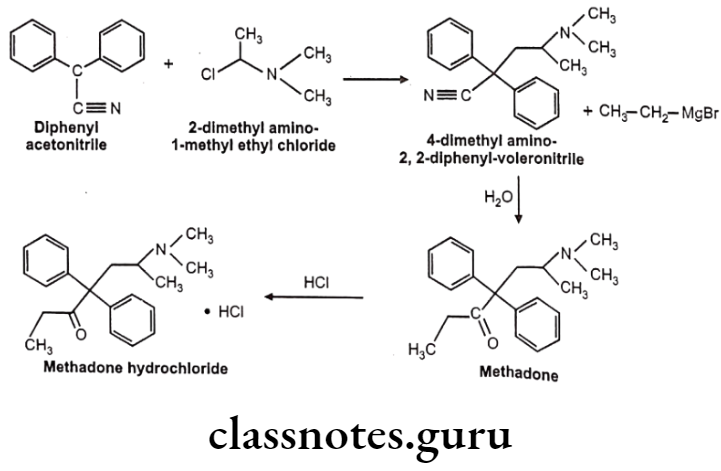
Methadone Hydrochloride:
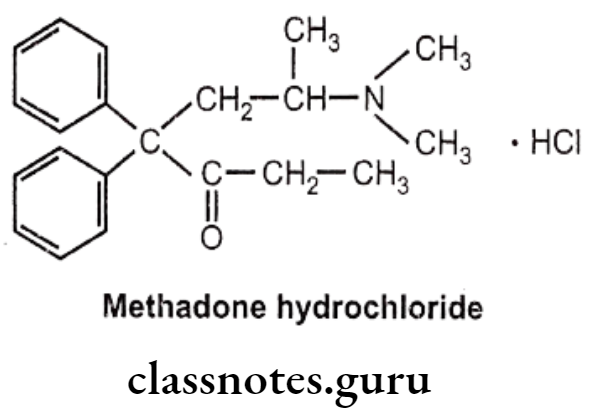
- Methadone hydrochloride is a synthetic narcotic drug.
- It is chemically, 6-(dimethylamino)-4,4-diphenylheptan-3-one.
- Methadone shows optical activity. Among the optical isomers, l-methadone is a more potent analgesic, while d-isomer is antitussive.
- It is more active and more toxic than morphine.
Uses:
- Methadone hydrochloride is used for the relief of many types of pain.
- It is also used in the treatment of some heroin addicts.
- It is used as a narcotic substitute in addiction treatment because it prevents morphine abstinence syndrome.
Propoxyphene Hydrochloride:
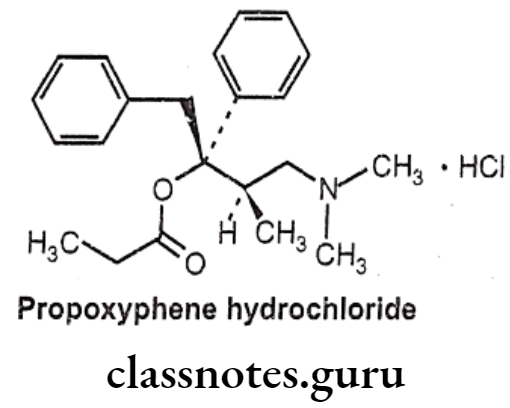
- Propoxyphene hydrochloride is chemically, [(25,3R)-4-(dimethylamino)-3-methyl-1,2- diphenylbutan-2-yl] propanoate; hydrochloride.
- This agent mimics the effects of the endogenous opiate dextropropoxyphene, by binding to μ-receptors located throughout the central nervous system.
Uses:
- Propoxyphene hydrochloride is used as narcotic analgesic.
Pentazocine:
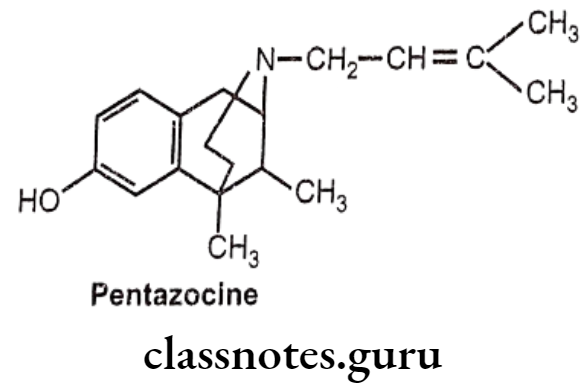
- Pentazocine is a novel drug possessing of both opioid agonistic and antagonistic properties.
- It is an agonist at the 8 and K opioid receptors and has a weak antagonist action at the μ-receptor.
- It presumably acts on K-receptors to produce analgesia and sedation.
- Like other narcotics, it produces analgesia, sedation and respiratory depression.
- The bioavailability of pentazocine after oral administration is only 20-50% due to the first pass metabolism.
- It gets metabolized extensively in the liver; and subsequently, excreted by the urinary tract.
Uses:
- Pentazocine is used in oral and parenteral forms as an analgesic for moderate-to- severe pain.
Levorphanol Tartarate:
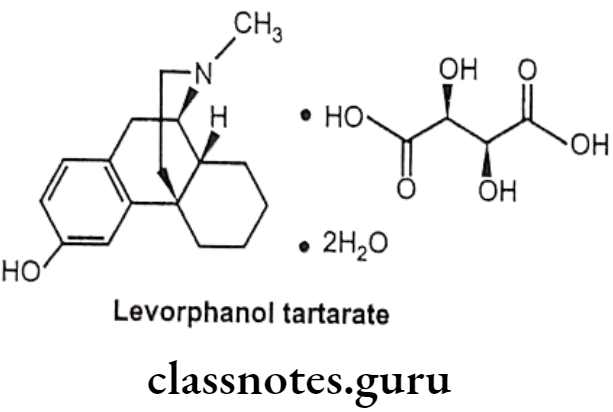
- Levorphanol tartarate is a synthetic phenanthrene with potent opioid analgesic activity.
- It mimics the actions of endogenous peptides at CNS opioid receptors, thereby producing the characteristic morphine-like effects on the μ-opioid receptor.
Uses:
- Levorphanol tartarate has morphine like effects such as analgesia, euphoria, sedation, respiratory depression, miosis, bradycardia and physical dependence.
Narcotic Antagonists
- Narcotic antagonists are drugs which block the “high” and other effects of narcotics. They also precipitate withdrawal symptoms in the narcotic addict.
- This feature of narcotic antagonists makes them extremely useful in treating overdoses.
- They are structurally related to morphine with the exception of the group attached to nitrogen, hence they act by competing for the same analgesic receptor sites. Research is currently going on to determine the usefulness of antagonists as maintenance drugs.
- Present narcotic antagonists (such as naloxone and cyclazocine) have too brief effect and too many side effects to be completely satisfactory.
- A new drug, naltrexone, appears to be more promising since its effects last longer, and it appears to be more acceptable to the treatment clients.
- Narcotic antagonists prevent or abolish excessive respiratory depression caused by the administration of morphine or related compounds.
- They are also used to treat asphyxia neonatorum and for the diagnosis of possible narcotic addiction.
Nalorphine Hydrochloride:
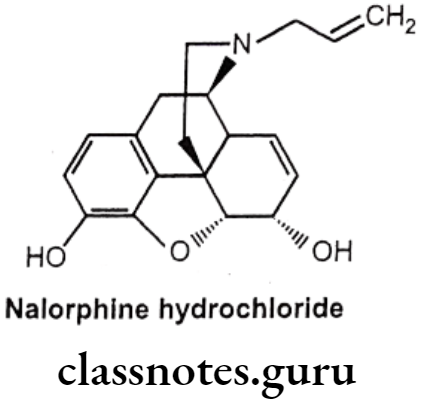
- Nalorphine hydrocholoride is N-allylmorphine. In morphine tertiary nitrogen is attached to an allyl (-CH2CH=CH2) group.
- It is a narcotic antagonist with some agonist properties. It is an antagonist at μ-opioid receptors and an agonist at K-opioid receptors.
- Nalorphine hydrochloride is white colored, odorless, crystalline powder.
- It darkens on exposure to light.
- It is soluble in water and dilute alkali hydroxide solution, but insoluble in chloroform and ether.
- It must be kept in tightly closed light resistant containers.
Uses:
- Nalorphine is a narcotic antagonist used to treat narcotic-induced respiratory depression.
- It is administered by intravenous injection for treating the overdosage of morphine, pethidine, methadone and levorphanol.
- Nalorphine precipitates withdrawal symptoms and produces behavioral disturbances in addition to the antagonism action.
Levallorphan Tartarate:
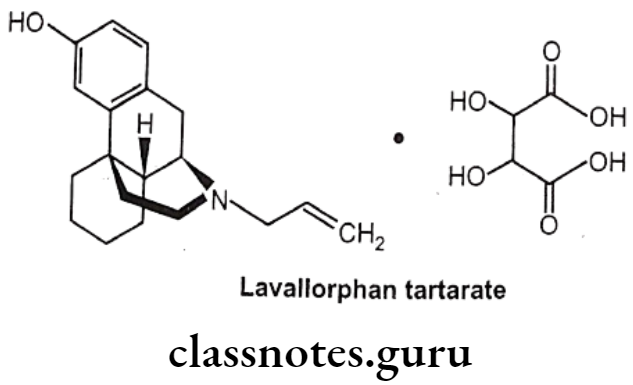
- Levallorphan is available as tartarate salt.
- Levallorphan tartarate occurs as white colored, odorless, crystalline powder.
- It melts at 175°C and is slightly soluble in water, but insoluble in ether and chloroform.
Uses:
- Levallorphan is a potent narcotic antagonist used in the treatment of narcotic induced respiratory depression.
Naloxone Hydrochloride:
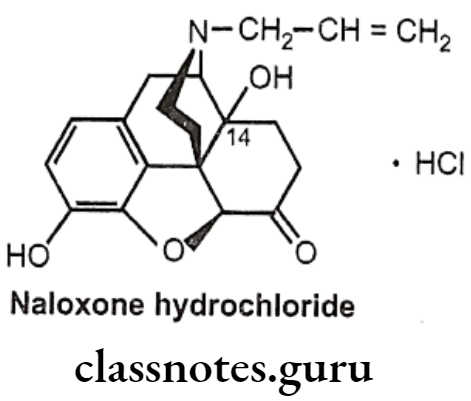
- Naloxone is (5R, 9R, 135, 14S)-N-allyl-4, 5-epoxy-3, 14-dihydroxymorphinan-6-one.
- It is a derivative of 7, 8-dihydro-14-hydroxymorphinone having an allyl group at the nitrogen.
- It is administered by IV or IM (low oral bioavailability and slow action) and has a relatively short half-life (1 hour).
- It is a specific narcotic antagonist which, unlike nalorphine, possesses no morphine- like properties. It is considered to be an effective antagonist for mixed agonist- antagonist like pentazocine.
- It may also reverse some of the adverse effects of narcotic antagonists having agonist actions owing to its lack of respiratory depressant property.
- It has been found to reverse narcotic analgesic and possesses little analgesic properties of its own.
Uses:
- Naloxone is a pure antagonist with no morphine like effects.
- It blocks the euphoric effect of heroin when given before heroin.
Synthesis
Fentanyl Citrate
Methadone Hydrochloride
Multiple Choice Questions:
Question 1. The drug naloxone
- produces morphine like activity
- produces respiratory depression
- induces constipation
- precipitates withdrawal symptoms in morphine addicts
Answer. 1. produces morphine like activity
Question 2. Kappa and delta agonists:
- inhibit postsynaptic neurons by opening K+ channels
- close a voltage-gated Ca** channels on presynaptic nerve terminals
- both (a) and (b)
- Inhibit of arachidonate cyclooxygenase in CNS
Answer. 2. close a voltage-gated Ca** channels on presynaptic nerve terminals
Question 3. Pentazocine is a benzomorphan derivative. It has alkyl group at C-3 position, identify it
- – CH = (CH3)2
- -CH2-CH=C(CH3)2
- = CH2-CH2-CH(CH3)2
- – CH(CH3)2
Answer. 2. -CH2-CH=C(CH3)2
Question 4. 3-etherification of morphine molecule causes
- Morphine antagonism
- No change in activity
- Decrease of analgesic and addiction
- Increase of analgesic and addiction.
Answer. 3. Decrease of analgesic and addiction
Question 5. Naltrexone is morphine
- Agonist
- Antagonist
- Partial antagonist
- All of above
Answer. 2.Antagonist
Question 6. The activity of the one of the following drug is dependant on phenyl-N-alkyl piperidine moiety:
- Meperidine
- Imipramine
- Diazepam
- Chlorpromazine
Answer. 1. Meperidine
Question 7. Which of the following analgesics is a phenanthrene derivative?
- Fentanyl
- Morphine
- Methadone
- Pentazocine
Answer. 2. Morphine
Question 8. Pentazocine belongs to the class
- Benzomorphans
- Morphinans
- Phenyl piperidine
- Azepine
Answer. 1. Benzomorphans
Question 9. Which one is the pure opioid antagonist among the following?
- Nalorphine
- Nalbuphine
- Naloxone
- Levallorphan
Answer. 3. Naloxone
Question 10. Tick narcotic analgesic, which is a phenylpiperidine derivative:
- Codeine
- Dezocine
- Fentanyl
- Buprenorphine
Answer. 3. Fentanyl
Question 11. Petazocine belongs to
- N-methylmorphinan
- Benzomorphan
- Meperidine
- Methadone
Answer. 2. Benzomorphan
Question 12. Morphine and heroin differs from each other in respect of
- Methyl group on nitrogen
- Acetyl group at C3 and C6
- Absence of double bond between C4 and C5
- Absence of D-ring
Answer. 2. Acetyl group at C3 and C6
Question 13. N,N-Dimethyl-(1-methyl-1-oxo-3,3-diphenyl hexyl) ammonium chloride is the chemical name for
- Methadone hydrochloride
- Meperidine hydrochloride
- Alpha proline hydrochloride
- Darvon
Answer. 1. Methadone hydrochloride
Question 14. Which of the following opioid analgesics is a strong μ-receptor agonist?
- Naloxone
- Morphine
- Pentazocine
- Buprenorphine
Answer. 2. Morphine
Question 15. Indicate the narcotic analgesic, which is a natural agonist:
- Meperidine
- Fentanyl
- Morphine
- Naloxone
Answer. 3. Morphine
Question 16. Select the narcotic analgesic, which is an antagonist or partial μ-receptor agonist:
- Fentanyl
- Pentazocine
- Codeine
- Methadone
Answer. 2. Pentazocine
Question 17. Which of the following agents is a full antagonist of opioid receptors?
- Meperidine
- Buprenorphine
- Naloxone
- Butorphanol
Answer. 3. Naloxone
Question 18. Which of the following opioid analgesics is used in obstetric labor?
- Fentanyl
- Pentazocine
- Meperidine
- Buprenorphine
Answer. 3. Meperidine
Question 19. Which of the following opioid agents is used in the treatment of acute opioid overdose?
- Pentazocine
- Methadone
- Naloxone.
- Remifentanyl
Answer. 3. Naloxone.
Anti-Inflammatory Agents
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are successfully used to treat a wide range of painful conditions and for the management of edema and tissue damage resulting from inflammation. Most of the drugs under this category are used in the treatment of fever as they possess antipyretic activity in addition to having analgesic and anti-inflammatory actions. Non-steroidal anti-inflammatory drugs (NSAIDs) reduce inflammation which helps to ease joint pain and stiffness. They are used in rheumatoid arthritis, osteoarthritis, ankylosing spondylitis and dysmenorrhea therapy.
Inflammation
The inflammatory response involves the migration of immune system cells into a damaged tissue. In some cases, this is beneficial especially for fighting infection, but in many cases, the inflammatory response increases the damage to the tissue such as in case of asthma, several forms of arthritis. In addition to this, inflammation may also be a step on the pathway towards certain cancers, especially colon cancer.
There are four signs of inflammation:
- Redness: Due to local vessel dilatation.
- Heat: Due to local vessel dilatation.
- Swelling: Due to influx of plasma proteins and phagocytic cells into the tissue spaces.
- Pain: Due to local release of enzymes and increased tissue pressure.
Role of Cyclooxygenase (COX):
Humans and most other mammals have two genes (isoenzyme) for cyclooxygenase, COX-1 and COX-2. Both are quite similar in structure, with only subtle differences. They catalyze the same reactions, although COX-2 works with a wider range of substrates.
COX-1 is constitutively expressed in most of the tissues such as gastrointestinal (GI) tract, the kidneys, and the circulatory system. In contrast, COX-2 is inducible, especially by inflammatory stimuli and found in few cell types such as macrophages, leukocytes, fibroblasts and endothelial cells.
COX-1 is responsible for generating the prostaglandins required for protection of the gastrointestinal tract, platelet aggregation, renal electrolyte haemostasis and renal blood flow maintanace while COX-2 is responsible for the increased prostaglandin synthesis associated with inflammation, fever, and pain responses.
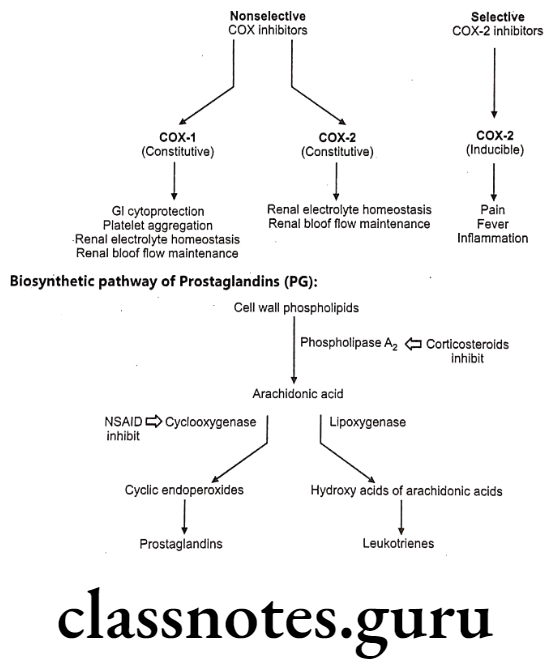
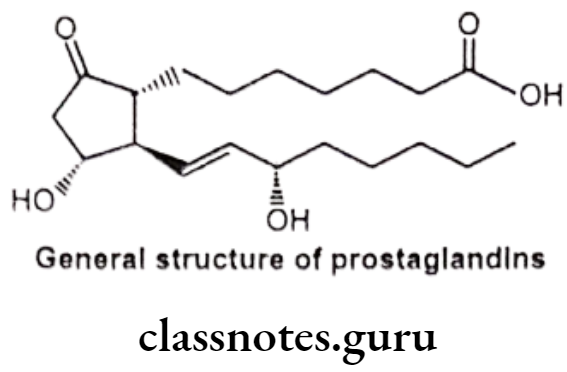
General Structure of Prostaglandins (PG):
Prostaglandins and related molecules are called eicosanoids. The term eicosanoid is derived from “eicosa” meaning “twenty”, referring to the 20 carbons in most of the molecules. Prostaglandin is a naturally occurring 20-carbon cyclopentano fatty acid derivative. Most prostaglandins are synthesized from arachidonic acid.
Mechanism of Action of NSAIDs:
Inflammation can be treated with two major classes of anti-inflammatory drugs: steroids (corticosteroids), and non-steroids. The steroids are compounds with glucocorticoid activity, and include the physiological glucocorticoid, cortisol, and synthetic glucocorticoid analogs such dexamethasone.
Glucocorticoids inhibit inflammatory responses by several mechanisms, and are more powerful drugs than NSAIDs. One mechanism is phospholipase A2 inhibition; this inhibits both prostaglandin and leukotriene synthesis, and therefore has a stronger effect than COX inhibition alone. In addition, glucocorticoids have other effects, unrelated to eicosanoid pathways.
The non-steroidal compounds are called Non-Steroidal Anti-Inflammatory Drugs (NSAIDs). Most NSAIDs inhibit both COX-1 and COX-2 with varying degree of selectivity. Selective COX-2 inhibitor may eliminate the side effects associated with NSAIDs due to COX-1 inhibition, such as gastric and renal effect. Some of the most widely used drugs, including aspirin, ibuprofen, and naproxen fall into this class.
Possible side-effects of NSAIDs include
- Stomach upsets
- Heartburn
- Indigestion
- Rashes
- Headaches
- Wheeziness
- Fluid retention, which can cause swelling of the ankles.
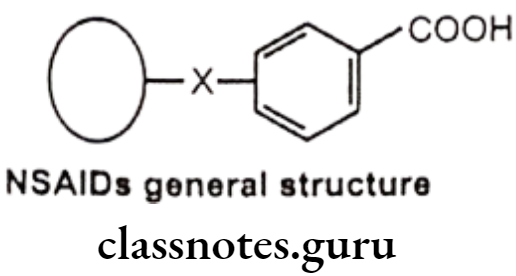
General Structure And Properties Of Nsaids
In general, NSAIDs structurally consist of an acidic moiety (carboxylic acid, enols) attached to a planar, aromatic functionality. Some analgesics also contain a polar linking group, which attaches the planar moiety to an additional lipophilic group. This can be represented as follows:
The NSAIDs are characterized by the following chemical/pharmacological properties:
- All are relatively strong organic acids with pKa in the 3.0-5.0 range. Most, but not all, are carboxylic acids. Thus, salt forms can be generated upon treatment with base and all of these compounds are extensively ionized at physiologic pH. The acidic group is essential for COX inhibitory activity.
- The NSAIDs differ in their lipophilicities based on the lipophilic character of their aryl groups and additional lipophilic moieties and substituents.
- The acidic group in these compounds serves a major binding group (ionic binding) with plasma proteins. Thus all NSAIDS are highly bound by plasma proteins (drug interactions).
- The acidic group also serves as a major site of metabolism by conjugation. Thus a major pathway of clearance for many NSAIDS is glucuronidation (and inactivation) followed by renal elimination.
Classification Of Anti-Inflammatory Agents
- Salicylic acid derivatives: Sodium salicylate, Aspirin, Diflunisal, Salsalate, Sulphasalazine.
- p-Amino phenol derivatives: Paracetamol, Phenacetin.
- Pyrazolidine dione derivatives: Phenyl butazone, Oxyphenbutazone, Sulphin- pyrazone.
- Anthranilic acid derivatives: Mefenemic acid, Flufenemic acid, Meclofenamate.
- Arylalkanoic acid derivatives:
- Indole acetic acid derivatives: Indomethacin.
- Indene acetic acid derivatives: Sulindac.
- Pyrrole acetic acid derivatives: Tolmetin, Zomepirac.
- Aryl and heteroaryl acetic/propionic acid derivatives: Ibuprofen, Diclofenac, Naproxen, Caprofen, Fenoprofen, Keto-profen, Flurbiprofen, Ketorolac, Etodaolac.
- Oxicams: Piroxicam, Meloxicam, Tenoxicam.
- Selective COX-2 inhibitors: Celecoxib, Rofecoxib, Valdecoxib.
- Miscellaneous: Nimesulide.
Salicylic Acid Derivatives (Salicylates)
- Salicylates have potent anti-inflammatory activity with mild analgesic and antipyretic activities.
- These compounds mainly act on COX-1 and bind higher affinity to COX-1.
- The therapeutic and some of the toxic actions of salicylates (aspirin) are related to its ability to inhibit COX-1 in various tissues and participate in trans-acetylation reactions in vitro.
Metabolism of salicylic acid derivatives: The initial route of metabolism of these derivatives is their conversion to salicylic acid, which is excreted in urine as free acid (10%) or undergoes conjugation with either glycine to produce the major metabolites of salicylic acid (75%) or with glucuronic acid to form glucuronide (15%).
In addition, small amount of metabolites resulting from microsomal aromatic hydroxylation leads to gentisic acid.
Structural – Activity Relationship (SAR) of Salicylates:
- The active moiety of salicylates is salicylate anion, side effects of aspirin, particularly GIT effects appear to be associated with the carboxylic acid functional group.
- Reducing the acidity of the carboxy group results in a change in the potency. of activity. Example: The corresponding amides (salicylamide) retains the analgesic action of salicylic acid, but is devoid of anti-inflammatory properties.
- Substitution on either the carboxyl or phenolic hydroxyl group may affect the potency and toxicity. Benzoic acid itself has only week activity.
- Placement of the phenolic hydroxyl group at meta or para position to the carboxyl group abolish the activity.
Sodium Salicylate:
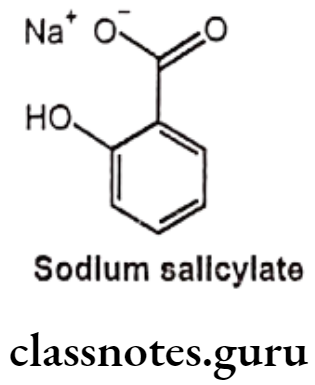
- Sodium salicylate is the sodium salt of salicylic acid.
- Sodium salicylate is chemically sodium; 2-hydroxybenzoate.
- It is an organic molecular entity.
- It irreversibly acetylates COX-1 and COX-2, thereby inhibiting prostaglandin synthesis and associated inflammation and pain.
- This salicylate produces the same adverse reactions as Aspirin, but there is less occult gastrointestinal bleeding.
Uses:
- Sodium salicylate is a non-steroidal anti-inflammatory agent, used to treat inflammation and pain.
Aspirin:
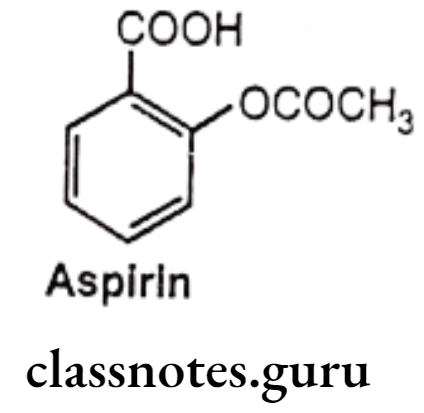
- Acetylsalicylic acid (aspirin) is an acetyl derivative of salicylic acid.
- It is chemically 2-acetyloxybenzoic acid.
- It acts as an inhibitor of cyclooxygenase which results in the inhibition of the biosynthesis of prostaglandins.
- It binds to and acetylates serine residues in cyclooxygenase.
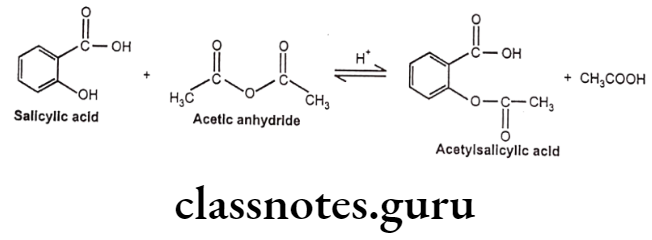
- It can be prepared by the reaction between salicylic acid and acetic anhydride. In this reaction, the hydroxyl group on the benzene ring in salicylic acid reacts with acetic anhydride to form an ester functional group. Thus, the formation of acetyl salicylic acid is referred to as an esterification reaction.
Uses:
- Aspirin is the prototypical analgesic used in the treatment of mild to moderate pain.
- It has anti-inflammatory and antipyretic properties.
- It also acts as antirheumatic.
- It also inhibits platelet aggregation and is used in the prevention of arterial and venous thrombosis.
p-Amino Phenol Derivatives (Anilides)
- These drugs are having somewhat different mechanism of action than other NSAIDs.
- They are believed to act as scavengers of hydroperoxide radicals (Hydroperoxide radicals have a stimulating effect on COX).
- Therefore the anilides have no anti-inflammatory action.
- The lack of an acidic functionality and COX inhibitory activity imparts several advantages including limited gastric irritation, ulceration, respiratory effects and little effect on platelets.
Metabolism of p-aminophenol Derivatives:
These drugs undergo hydrolysis to yield aniline derivatives that produce directly or through their conversion to hydroxylamine derivatives, such as acetaminophen that undergoes rapid first pass metabolism in the GIT to o-sulphate conjugate.
The N-hydroxylamine is then converted into a reactive toxic metabolite, acetimino- quinone, which produces toxicity to the kidney and liver in conjugation with hepatic glutathione to form mercapturic acid or cysteine conjugates.
Structure – Activity Relationship of p-amino Phenol Derivatives:
- Etherification of the phenolic function with methyl or propyl groups produces derivatives with greater side effects than ethyl derivatives.
- Substituents of the nitrogen atom, which reduce the basicity, also reduce activity unless the substituent is metabolically labile. e.g. acetyl groups.
- Amides derived from aromatic acid. e.g. N-phenyl benzamides that are less active or inactive.
Phenacetin (Acetophenetidin):
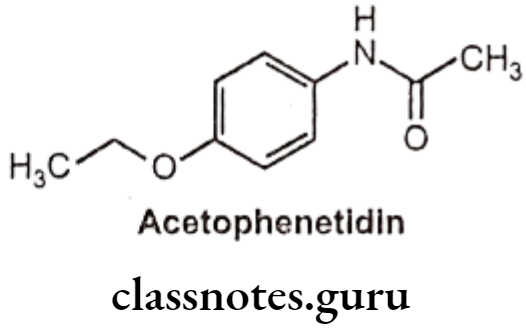
- Phenacetin is chemically N-(4-ethoxyphenyl) acetamide.
- It is a phenylacetamide that was formerly used in analgesics, but nephropathy and methemoglobinemia led to its withdrawal from the market.
Uses:
- Phenacetin is an analgesic and an antipyretic with similar effectiveness as an aspirin.
- It has a greater potential for toxicity (hemolytic anaemia and methemoglobinaemia) than paracetamol.
Paracetamol (p-acetaminophen):
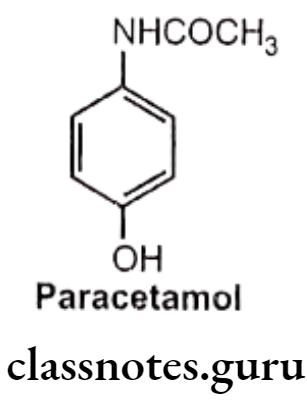
- Paracetamol is chemically N-(4-hydroxyphenyl)acetamide.
- Paracetamols produce antipyresis by inhibition of prostaglandin synthesis and release in the central nervous system (CNS) and by acting on the hypothalamic heat- regulating centre.
- It produces analgesia by elevating the pain threshold.
- It may inhibit the nitric oxide (NO) pathway mediated by a variety of neurotransmitter receptors including N-methyl-D-aspartate (NMDA), resulting in elevation of the pain threshold.
- Hepatic necrosis and death have been observed following over dosage.
- It may cause liver, blood cell, and kidney damage.
Uses:
- Acetaminophen is a p-aminophenol derivative with analgesic and antipyretic activities.
- Acetaminophen has weak anti-inflammatory properties and is used as a common analgesic.
3,5-Pyrazolidinedione Derivatives
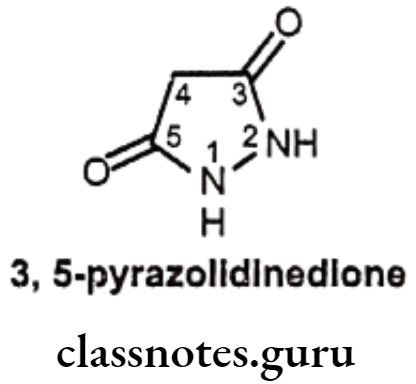
Structure – Activity Relationship of 3,5-Pyrazolidinediones:
- Replacement of one of the nitrogen atom in the pyrazolidinediones with an oxygen atom yields isoxazole analogues, which are as active as pyrazolidinedione derivatives.
- In 3,5-pyrazolidinedione derivatives, pharmacological activities are closely related to their acidity, the dicarbonyl function at the 3rd and 5th positions enhance the acidity of hydrogen atom at the 4th position.
- Presence of a keto group in the y-position of the butyl side chain produces the active compound.
- Decreasing or eliminating acidity by removing the acidic proton at 4th position (e.g. 4, 4-dialkyl derivatives) abolishes anti-inflammatory activity. Thus, if the hydrogen atom at the 4th position of phenyl butazone is replaced by substituents, such as a methyl group, anti-inflammation activity is abolished.
- If acidity is enhanced too much, anti-inflammatory and sodium-retaining activities decrease; while other properties, such as the uricosuric effect increase.
- Introduction of polar function in these alkyl groups give mixed results. The -hydroxy-n-butyl derivative possesses pronounced uricosuric activity, but give fewer anti-inflammatory effects.
- Substitution of 2-phenyl thio ethyl group at the 4th position produces antigout activity (sulphinpyrazone).
- Presence of both the phenyl groups is essential for neither anti-inflammatory nor analgesic activity.
- m-substitution of aryl rings of the phenyl butazone gives uniformly inactive compounds.
- p-substitution, such as methyl, chloro, nitro, or OH of one or both rings retains activity.
Phenylbutazone:
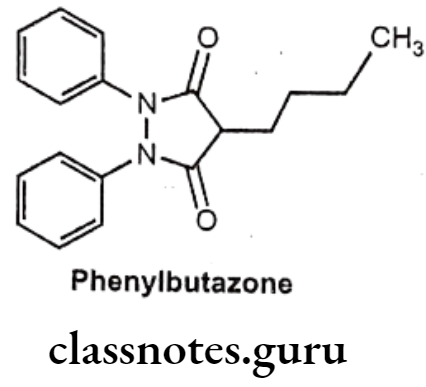
- Phenylbutazone is chemically, 4-butyl-1,2-diphenylpyrazolidine-3,5-dione.
Uses:
- Phenylbutazone is a pyrazole derivative that has antipyretic, analgesic, and anti- inflammatory actions.
- It is especially effective in the treatment of ankylosing spondylitis.
- It is also useful in arthritis, acute superficial thrombophlebitis, painful shoulder, and Reiter’s disease.
Antipyrine:
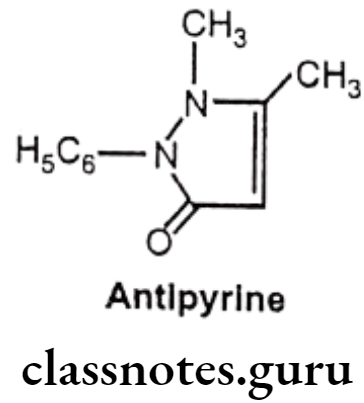
- Antipyrine is chemically, 2, 3-dimethyl-1-phenyl-3-pyrazolin-5-one.
Uses
- Antipyrine has analgesic, anti-inflammatory and antipyretic activities.
- It exerts a paralytic action on the sensory and the motor nerves, resulting in some anesthesia and vasoconstriction, and it also exerts a feeble antiseptic effect.
Anthranilic Acid Derivatives (Fenamates)
- The anthranilates are primarily anti-inflammatory with some analgesic and antipyretic activity and are non-COX selective.
- The anthranilates are used as mild analgesics and occasionally to treat inflammatory disorders.
- Diclofenac is used for rheumatoid arthritis, osteoarthritis and post-operative pain and mefenamic acid as an analgesic for dysmennorhea.
- The utility of this class of agents is limited by a number of adverse reactions including nausea, vomiting, diarrhoea, ulceration, headache, drowsiness and hematopoietic toxicity.
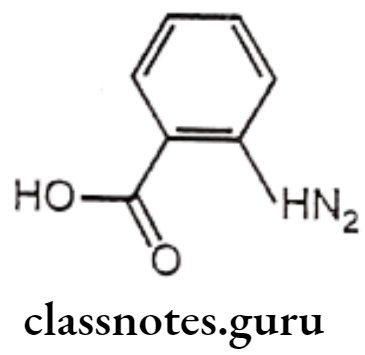
Structure – Activity Relationship of Anthranilic Acid Derivatives (Fenamates):
- The position of the carboxyl function is important for the activity of anthranilic acid derivatives that are active, whereas the 3 and 4 amino benzoic acid analogues are not active.
- Replacement of carboxylic acid function with the isosteric tetrazole results in the retention of anti-inflammatory activity.
- Placement of substitution on the anthranilic acid ring generally reduces the activity.
- Substitution on the N-aryl ring can lead to conflicting results. In the ultraviolet erythema assay for anti-inflammatory activity, the order of activity was generally 3′> 2′>4′ for mono-substitution with CF3 group (flufenamic acid) being particularly potent. The opposite order of activity was observed in rat paw oedema assay, the 2′-Cl derivatives being more potent than 3′-Cl analogues.
- In disubstituted derivatives, where the nature of the two substitutes is the same, 2′,3′-disubstitution appears to be the most effective (mefenemic acid).
- The NH moiety of anthranilic acid is essential for the activity as the replacement of NH function with O, CH2, S, SO2, N-CH3, or NCOCH3 functionalities significantly reduced the activity.
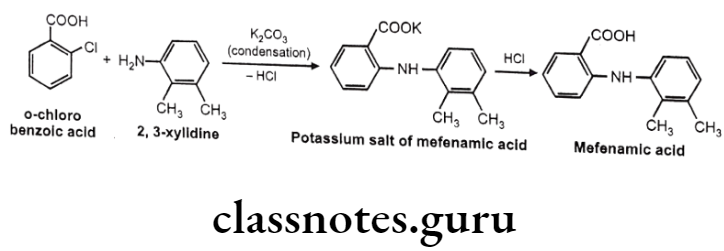
Mefenamic acid:
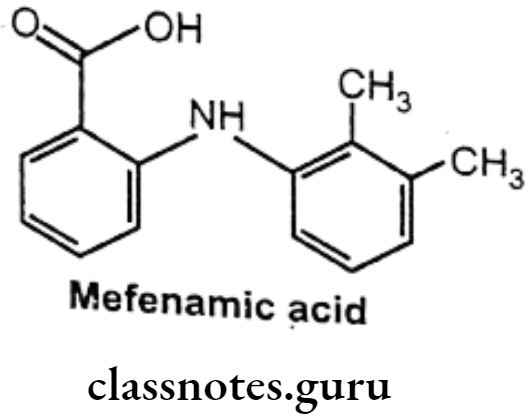
- Mefenamic acid is anthralinic acid NSAID.
- It is chemically 2-(2,3-dimethylphenylamino) benzoic acid.
- It is an inhibitor of cyclooxygenase.
- It inhibits the activity of the enzyme COX-1 and COX-2, resulting in a decreased formation of precursors of prostaglandins and thromboxanes, thereby inhibiting platelets aggregation.
Uses:
- Mefenamic acid is used as an analgesic and anti-inflammatory agent.
- It also possesses antipyretic property.
Metabolism:
- Its metabolism occurs through regioselective oxidation of 3-methyl group and glucuronidation of mephanamic acid. Majority of the 3-hydroxy methyl metabolites and dicarboxylic acid products are excreted.
Meclofenamate Sodium:
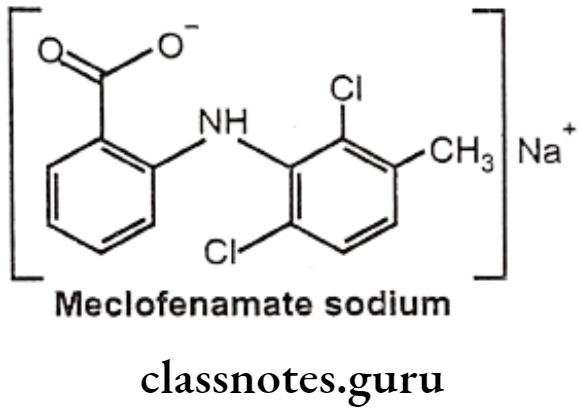
- Meclofenamate sodium is anthralinic acid NSAID.
- It is chemically, sodium 2-(2,6-dichloro-3-methylphenylamino)benzoate.
- It inhibits the activity of the enzyme COX-1 and COX-2, resulting in a decreased formation of precursors of prostaglandins and thromboxanes thereby inhibiting platelets aggregation.
Uses:
- Meclofenamate sodium is used as an analgesic and anti-inflammatory agent.
- It also possesses antipyretic property.
Arylalkanoic Acids
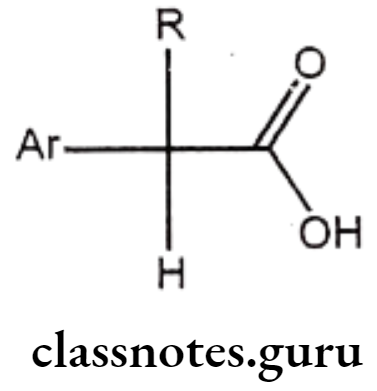
Structure – Activity Relationship of Arylalkanoic Acids:
- The largest group of NSAIDS is represented by the class of arylalkanoic acids.
- Drugs of this class share a number of common structural features.
- The centre of acidity is usually located one carbon atom adjacent to a flat surface represented by an aromatic or hetero aromatic ring.
- The distance between these centres is crucial, because increasing this distance to two or three carbons generally decreases activity.
- All agents possess a centre of acidity, which can be represented by a carboxylic acid (R=COOH) and hydroxamic acid (R=CONHOH), a sulphonamide (R=SO2NH2), phenol or a terazole.
- Substitution of a methyl group on the carbon atom separating the aromatic ring leads to enhancement of anti-inflammatory activity.
- Groups larger than methyl decrease activity, but incorporation of this methyl group as part of an alicyclic ring system does not drastically affect activity.
Indole Acetic Acid Derivatives:
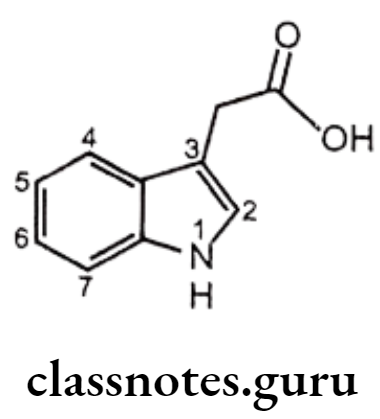
Structure – Activity Relationship of Indene and Indole Acetic Acid Derivatives:
- The carboxyl group is essential for anti-inflammatory activity.
- Placement of other acidic functionalities instead of the carboxyl group decreases activity and the amide derivatives are inactive.
- Alkyl group especially -CH3 at 2nd position is much active than aryl substituted analogues.
- The 5th position of the indole ring is most flexible with regard to the nature of substituents that enhance activity. Substituents such as methoxy, fluoro, dimethylamino, methyl, allyloxy, and acetyl are more active than the unsubstituted indole ring.
- The presence of indole ring nitrogen is not essential for activity because the corresponding 1-benzylidenylindene analogue (sulindac) is also active.
- Acylation of the indole nitrogen with aryl/alkyl carboxylic acids results in the decrease of activity.
- Presence of substituents on the N-benzoyl derivatives in the p-position with F, CI, CF3, or S-CH3 groups provide greatest activity.
Indomethacin:
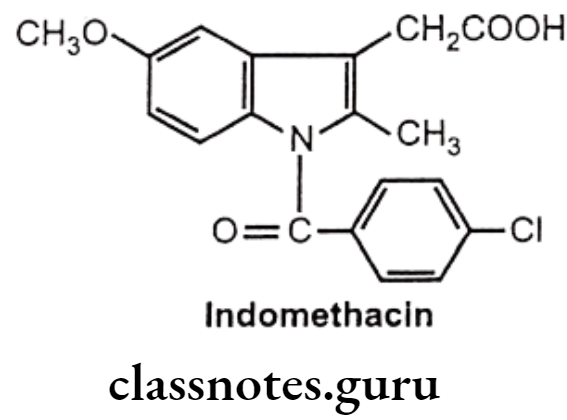
- Indomethacin is chemically, 1-(p-chloro benzoyl)-5-methoxy-2-methylindole-3-acetic acid.
- It inhibits the enzyme COX necessary for the formation of prostaglandins and other autocoids.
Uses:
- It is a more potent antipyretic than either aspirin or acetaminophen.
- It possesses approximately 10 times the analgesic potency of aspirin.
- It is used as anti-inflammatory and analgesic in rheumatic arthritis, spondylitis, and to lesser extent in gout.
- The most frequent side effects are gastric distress and headache.
- It has also been associated with peptic ulceration, blood disorders, and possible deaths.
- It is recommended only for patients who cannot tolerate aspirin and in place of phenylbutazone in long-term therapy.
Indene Acetic Acid Derivatives:
Sulindac:
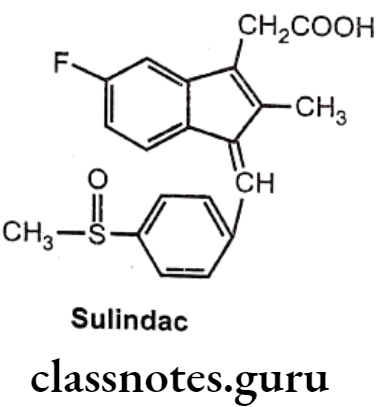
- Sulindac is a prodrug.
- It is chemically 5-fluoro-2-methyl-1[(4 methyl sulphinyl) phenyl methylene] indene-3- acetic acid.
Metabolism:
- Sulindac reaches peak blood levels within 2 to 4 hours and undergoes a complicated, reversible metabolism. The parent sulfinyl has a plasma half-life of 8 hours. It forms active metabolites of sulphide having plasma half-life of 16.4 hours. In addition to it, sulindac is oxidized to corresponding sulfoxide. The more polar and inactive sulphoxide is virtually the only form excreted.
Uses:
- Sulindac has analgesic, antipyretic, and anti-inflammatory properties.
- It is recommended for rheumatoid, osteoarthritis and ankylosing spondylitis.
- It is used in the treatment of muscular skeletal disorders and acute gouty arthritis.
- It may produce gastric bleeding, nausea, diarrhoea, dizziness as adverse effects, but with a lower frequency than with aspirin.
Pyrrole Acetic Acid Derivatives:
Structure – Activity Relationship of Pyrrole Acetic Acid Derivative:
- Replacement of the p-tolyl group (tolmetin) with a p-chloro benzoyl moiety produced little effect on activity.
- Introduction of a methyl group in the 4th position and 5-p-chloro benzoyl analogues (zomepirac) proved to be four times potent as tolmetin.
Tolmetin Sodium:
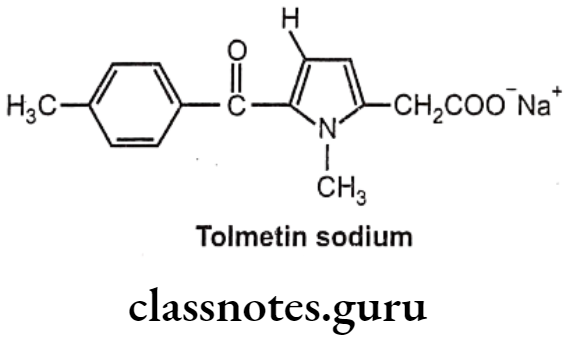
- Tolmetin sodium is chemically, sodium-2-(1-methyl-5-(4-methylbenzoyl)-1H-pyrrol- 2-yl) acetate.
- It inhibits the enzyme prostaglandin synthase, thus prevent synthesis of the inflammatory prostaglandin E2 (PGE2) from the precursor prostaglandin H2 (PGH2).
Uses:
- Tolmetin sodium has antipyretic, analgesic, and anti-inflammatory actions.
- It is employed in the treatment of rheumatic and musculoskeletal disorders.
- The drug is, however, comparable to indomethacin and aspirin in the control and management of disease activity.
Zomepirac:
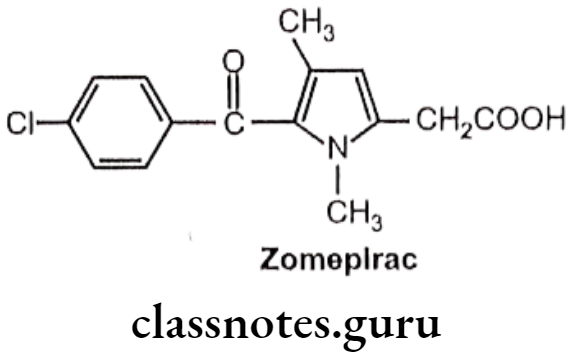
- Zomepirac is chemically, 1,4-dimethyl-5-(p-chloro benzoyl)pyrrole-2-acetic acid.
- It has associated with fatal and near-fatal anaphylactoid reactions.
Uses:
- Zomepirac is recommended in greater degree of analgesia for severe pain.
- It is used as an analgesic and an anti-inflammatory drug.
- It is four times as potent as tolmetin.
Aryl and Heteroaryl Acetic/Propionic Acid Derivatives:
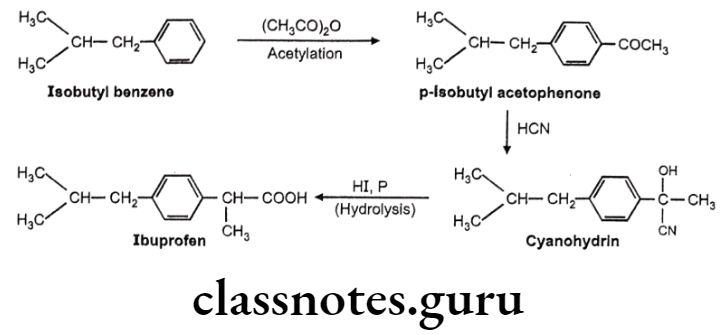
Ibuprofen:
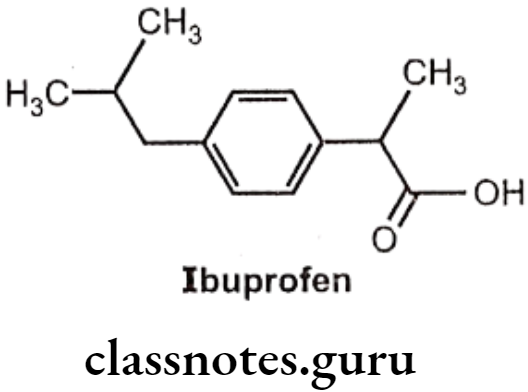
- Ibuprofen is chemically 2(p-isobutyl-phenyl)-propionic acid.
- The activity resides in the (s)-(+) isomer, not only in Ibuprofen, but also throughout the arylacetic acid series.
- These isomers are the more potent inhibitors of prostaglandin synthase.
- The precursor Ibufenac (acetic acid in place of propionic acid), has abandoned hepatotoxicity and is less potent.
Uses:
- Ibuprofen is an anti-inflammatory drug that possesses antipyretic and analgesic action and is used for the treatment of rheumatoid arthritis and osteoarthritis.
Diclofenac:
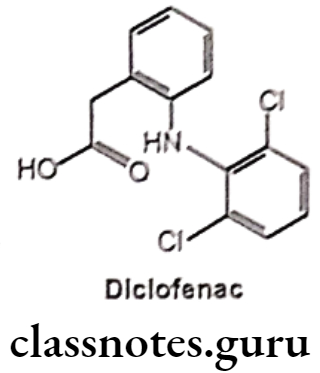
- Diclofenac is chemically o-(2,6-dichloro anilino)-phenyl acetic acid.
- It is available in sodium and potassium salt form.
- It acts by inhibiting COX and decreased prostaglandin production.
- It binds and chelates both COX-1 and COX-2, thereby blocking the conversion of arachidonic acid to pro-inflammatory-prostaglandins.
Uses:
- Diclofenac sodium is used in the treatment of rheumatic arthritis, osteoarthritis and ankylosing spondylitis.
- The potassium salt is faster acting and is used for the management of acute pain.
Naproxen:
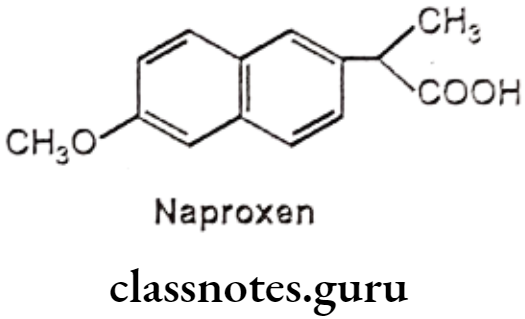
- Naproxen is chemically, (±) 2-(6-methoxy-2-naphthyl)-propionic acid.
- The effectiveness of naproxen is partly due to its ability to inhibit COX-1 and COX-2, resulting in a decreased formation of precursors of prostaglandins and thromboxanes.
- It decreases in the formation of thromboxane A2 synthesis, by thromboxane synthase, thereby inhibiting platelet aggregation.
- It irreversibly blocks the enzyme cyclooxygenase (prostaglandin synthase), which catalyzes the conversion of arachidonic acid to endoperoxide compounds.
Uses:
- Naproxen is fairly comparable to aspirin both in the management and control of disease symptoms.
- It has lesser frequency and severity of nervous system together with milder GI-effects. It possesses analgesic, anti-inflammatory, and antipyretic actions.
- It is used in the treatment of rheumatic arthritis, dysmenorrhea, and acute gout.
Ketorolac:
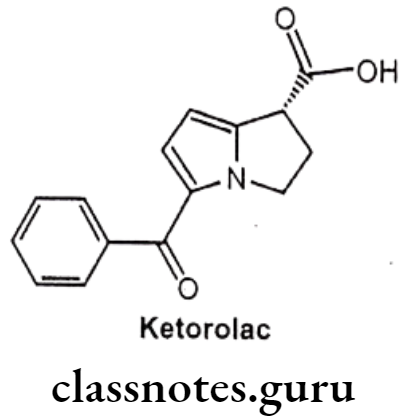
- Ketorolac is chemically 5-benzoyl-2,3-dihydro-1H-pyrrolizine-1-carboxylic acid.
- It is non-selective and it inhibits the enzymes COX-1 and COX-2.
- The inhibition of COX-2, prevents conversion of arachidonic acid to pro-inflammatory prostaglandins, whereas the inhibition of COX-1 prevents the normal steady-state production of prostaglandins that play housekeeping roles in the protection of the gastrointestinal tract.
Uses:
- Ketorolac is a potent analgesic indicated for the treatment of moderately severe and acute pain.
- It also possesses analgesic and antipyretic activities.
- Because of a number of potential side effects, its administration should not exceed 5 days.
Oxicams
- Oxicams are COX-2 selectivity than many other NSAIDs, particularly meloxicam.
- These agents have utility in treatment of rheumatoid arthritis and osteoarthritis.
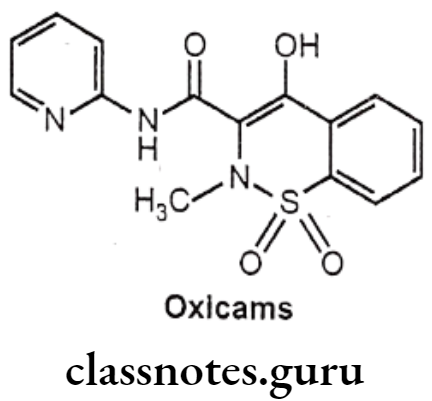
Structure Activity Relationship of Oxicams:
- The most active analogues have substituent CH3 on the nitrogen and electron withdrawing substituents on the anilide phenyl groups, such as – Cl and -CF3.
- The introduction of heterocyclic ring in the amide chain significantly increases the anti-inflammatory activity. Example: 2-thiazolyl derivative sudoxicam is more potent than indomethacin.
- The most active benzothiazine have acidities in the pKa range of 6-8.
Piroxicam:
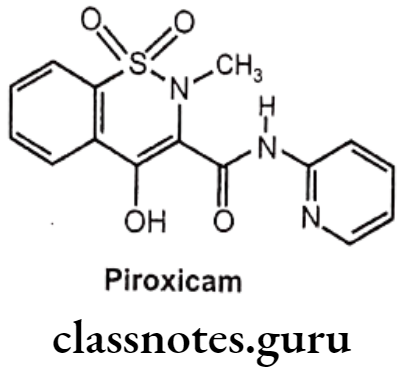
- Piroxicam is chemically, 4-hydroxy-2-methyl-N-2-pyridinyl-1,2-benzothiazine-3- carboxamide-1,1-dioxide.
- It binds and chelates both isoforms of COX-1 and COX-2 and prevent conversion of arachidonic acid into prostaglandin.
Uses:
- Piroxicam has anti-inflammatory, antipyretic and analgesic properties.
- It employed for acute and long-term therapy for the relief of symptoms of osteoarthritis and rheumatoid arthritis.
- It also possesses uricosuric action and has been used in the treatment of acute gout.
Selective COX-2 Inhibitors
- Cyclooxygenase-2 (COX-2) is an inducible enzyme release in response to injury or inflammation.
- COX-2 inhibitors are the new-generation NSAIDs that may selectively block the COX-2 isoenzyme without affecting COX-1 function.
- This may result in control of pain and inflammation with a lower rate of adverse effects compared with older non-selective NSAIDs.
- Rapidly evolving evidence suggests that COX-2 enzyme has a diverse physiologic and pathologic role.
Celecoxib:
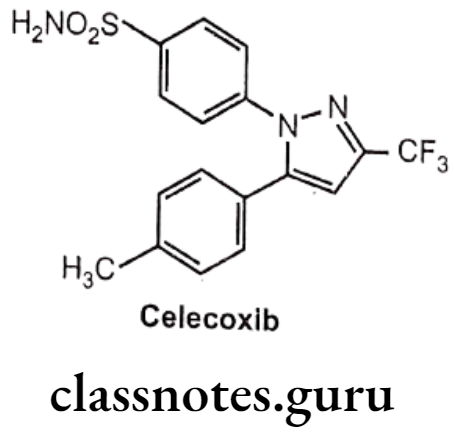
- Celecoxib is chemically, 4-[5-(4-methylphenyl)-3-(trifluoromethyl)pyrazole-1-yl]- benzenesulfonamide.
- It has a central pyrazole ring with two adjacent phenyl substituents.
- One phenyl ring contains a methyl group at p-position, whereas other contains a polar sulfonamide moiety at p-position.
- The sulfonamide have ability to bind with a distinct hydrophilic region that is present on COX-2, but not COX-1.
Rofecoxib
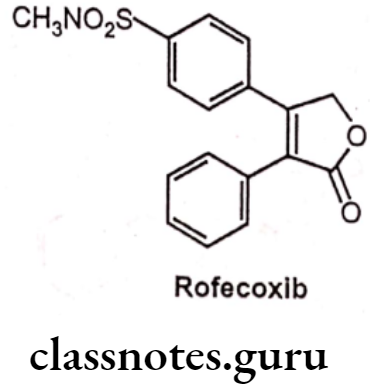
- Rofecoxib is chemically, 3-(4-methylsulfonylphenyl)-4-phenyl-2H-furan-5-one.
- It has a central furanose ring and two adjacent phenyl substituents.
- One phenyl ring contains a methyl sulfone group, whereas other phenyl ring is unsubstituted.
- It binds to and inhibits the enzyme COX-2 resulting in an inhibition of the conversion of arachidonic acid to prostaglandins.
- It has greater potency and a longer half-life than celecoxib.
Valdecoxib
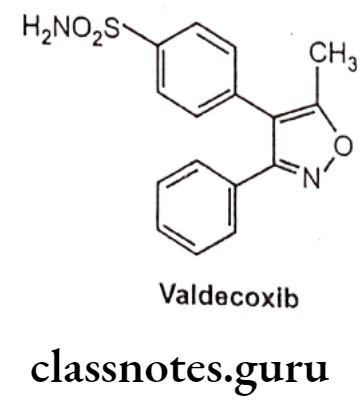
- Valdecoxib is chemically, 4-(5-methyl-3-phenyl-1,2-oxazol-4-yl)-benzenesulfonamide.
- It is an aryl sulfonamide derivative like celecoxib.
- It acts by inhibiting prostaglandin synthesis by blocking COX-2 thereby preventing the conversion of arachidonic acid to prostaglandins, which are involed in the regulation of pain, fever and inflammation.
- At therapeutic plasma concentrations valedecoxib does not inhibit COX-1.
Uses:
- Valdecoxib is non-steroidal anti-inflammatory drugs that exhibit anti-inflammatory, analgesic and antipyretic properties.
- These drugs are also recommended for rheumatoid arthritis, osteoarthritis and juvenile arthritis.
- It is also recommended to treat painful menstruation and menstrual symptoms.
Miscellaneous
Nimesulide:
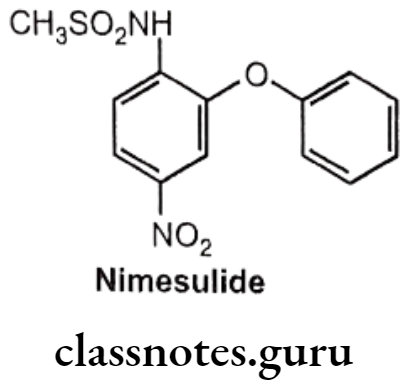
- Nimesulide is chemically N-(4-nitro-2-phenoxyphenyl)-methanesulfonamide.
- It inhibits the COX (COX-2) mediated conversion of arachidonic acid to pro- inflammatory prostaglandins.
Uses:
- Nimesulide has anti-inflammatory activity.
- It is recommended in the treatment of acute pain.
Synthesis
Mafenamic Acid
Ibuprofen
Multiple Choice Questions:
Question 1. Non-narcotic analgesics are mainly effective against pain associated with:
- Inflammation or tissue damage
- Trauma
- Myocardial infarction
- Surgery
Answer. 1. Inflammation or tissue damage
Question 2. IUPAC name of the sulindac analogue is
- (Z)-5-fluoro-2-methyl-1-[(p-methyl sulfinyl)phenyl] methylene-1H-indene-3- acetic acid
- (E)-5-fluoro-2-methyl-1-phenyl methylene-1H-indene-3-acetic acid
- (Z)-5-fluoro-2-methyl-2-[(p-methyl sulfinyl)phenyl] methylene-1H-indene-4- acetic acid
- (R)-5-fluoro-2-methyl-1-1-phenyl methylene-1H-indene-3-acetic acid
Answer. 1. (Z)-5-fluoro-2-methyl-1-[(p-methyl sulfinyl)phenyl] methylene-1H-indene-3- acetic acid
Question 3. Non-narcotic analgesics are all of the following drugs EXCEPT:
- Paracetamol
- Acetylsalicylic acid
- Butorphanol
- Ketorolac
Answer. 3. Butorphanol
Question 4. Which isomer of ibuprofen is more active?
- (S) (-) isomer
- (S) (+) isomer
- (R) (+) isomer
- (R) (-) isomer
Answer. 2. (S) (+) isomer
Question 5. Select the non-narcotic drug, which is a para-aminophenol derivative:
- Analgin
- Aspirin
- Baclophen
- Paracetamol
Answer. 4. Paracetamol
Question 6. Phenylbutazone is the acidic drug. It is due to
- Easily replaceable hydrogen
- CO-CH2-CO moiety
- Two keto groups
- Two nitrogen atoms
Answer. 2. CO-CH2-CO moiety
Question 7. Which of the following is an active form of sulindac?
- Z-form
- E-form
- Z & E-form
- None of above
Answer. 1. Z-form
Question 8. Which of the following non-narcotic agents is salicylic acid derivative?
- Phenylbutazone
- Ketamine
- Aspirin
- Tramadol
Answer. 3. Aspirin
Question 9. Starting material for ibuprofen is
- Isopropyl benzene
- Isobutyl benzene
- Isobutyl acetophenone
- Isopropyl acetophenone
Answer. 2. Isobutyl benzene
Question 10. Pyrazolone derivative is:
- Methyl salicylate
- Analgin
- Paracetamol
- Ketorolac
Answer. 2. Analgin
Question 11. Which of the following is N-aryl anthranilic acid derivative?
- Mefenamic acid
- Toletine
- Indomethacine
- Paracetamol
Answer. 1. Mefenamic acid
Question 12. Chemically ibuprofen is
- 2-(4-isobutyl phenyl) propionic acid
- 3,20-di-oxo-4-pregnen-17a-yl benzoate
- 2-(4-isopropyl phenyl) propionic acid
- 2-(4-isopropyl methyl) propionic acid
Answer. 1. 2-(4-isobutyl phenyl) propionic acid
Question 13. Which one of the following non-narcotic agents inhibits mainly cyclooxygenase (COX) in CNS?
- Paracetamol
- Ketorolac
- Acetylsalicylic acid
- Ibuprofen
Answer. 1. Paracetamol
Question 14. Which of the following ring is present in sulindac?
- Indol
- Indene
- Isoxazole
- Furan
Answer. 2. Indene
Question 15. Most of non-narcotic analgetics have:
- Anti-inflammatory effect
- Analgesic effect
- Antipyretic effect
- All of the above
Answer. 4. All of the above
Question 16. Indicate the non-narcotic analgesic, which lacks an anti-inflammatory effect:
- Naloxone
- Paracetamol
- Metamizole
- Aspirin
Answer. 2. Paracetamol
Question 17. Ibuprofen is a
- 2-(4-propylphenyl)propionic acid
- 2-(4-isobytylphenyl)propionic acid
- 2-(4-ethylphenyl)propionic acid
- 2-(4-hexylphenyl)propionic acid
Answer. 2. 2-(4-isobytylphenyl)propionic acid
Question 18. Paracetamol is a 4-acetaminophenol. It is an intermediate in the preparation of
- Chlorphenesin
- Phenacetine
- Hexachlorophan
- Hexylresorcinol
Answer. 2. Phenacetine
Question 19. Methemoglobinemia is possible adverse effect of:
- Aspirin
- Paracetamol
- Analgin
- Ketorolac
Answer. 2. Paracetamol