
Drug Design Introduction
Drug discovery is the process through which potential new therapeutic entities are identified, using a combination of computational, experimental, translational, and clinical models.
- Despite advances in biotechnology and understanding of biological systems, drug discovery is still a lengthy, costly, difficult, and inefficient process with a high attrition rate of new therapeutic discovery.
- Drug design is the inventive process of finding new medications based on the knowledge of a biological target.
- In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the molecular target with which they interact and bind.
- Drug design frequently but not necessarily relies on computer modeling techniques and bioinformatics approaches in the big data era.
- In addition to small molecules, biopharmaceuticals and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also gained great advances.
- Drug development and discovery includes preclinical research on cell-based and animal models and clinical trials on humans, and finally move forward to the step of obtaining regulatory approval to market the drug.
- Modern drug discovery involves the identification of screening hits, medicinal chemistry, and optimization of those hits to increase the affinity, selectivity (to reduce the potential of side effects), efficacy or potency, metabolic stability (to increase the half-life), and oral bioavailability.
- Once a compound that fulfills all of these requirements has been identified, it will begin the process of drug development before clinical trials.
Factors Affecting Drug Discovery
Several factors affect the drug discovery and development process.
Important Ones Are As Follows:
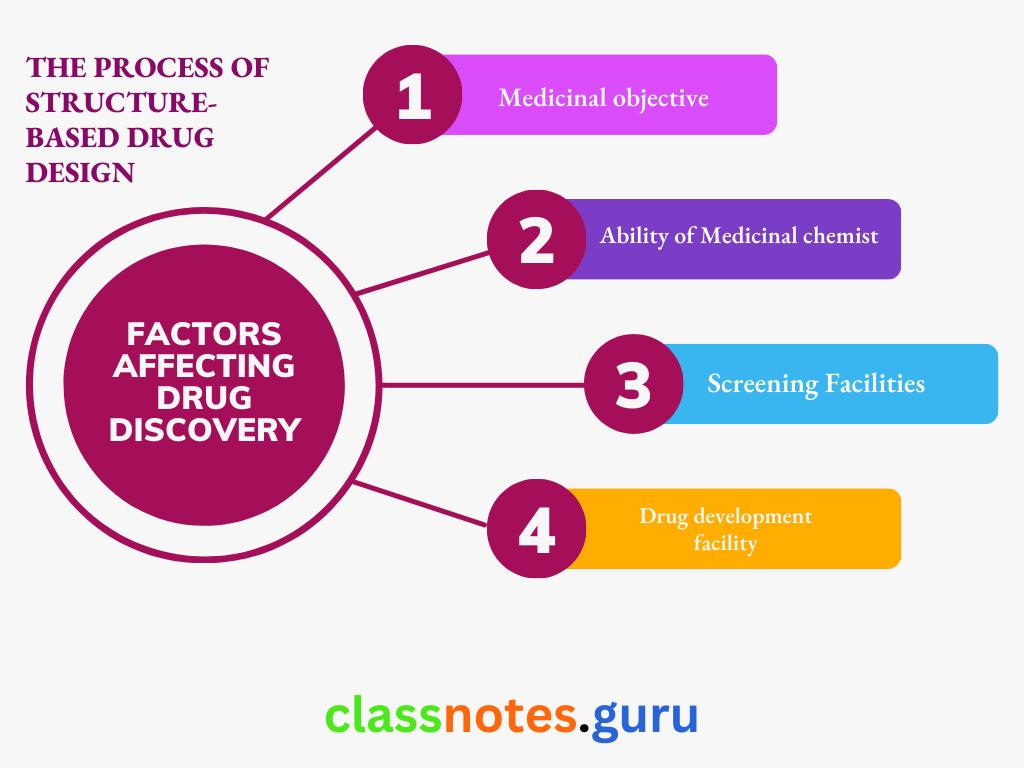
- Medicinal Objective: In general, the more precise the medicinal objective, the less likely it is to develop a new drug; for example, it is easy to develop an antacid but much more difficult to develop a specific proton pump inhibitor. Thus, the medicinal requirements affect the likelihood of success or failure in new drug discovery.
- Ability Of Medicinal Chemist: The attributes of the chemist will influence the outcome of evolving new drugs based on knowledge of the chemistry of lead molecules and the biology of a diseased state.
- Screening Facilities: A successful and rapid mass screening mainly depends on the capacity to evaluate a large number of compounds and detect potentially clinically useful drugs in a very short period.
- Drug Development facility: Good facilities with interdisciplinary efforts by chemistry, biology, pharmacy, and medical groups are necessary for drug development.
Read and Learn More Medicinal Chemistry III Notes
Cost Of New Drug
The following three factors affect the cost of drug development
- Number Of Compounds Synthesized: Of the about 5000-10,000 compounds studied, only one drug reaches the market.
- Nature of the lead molecule: The cost of production will be high if the lead molecule is prepared by an expensive route.
- Standards Required For New Drugs: The standards required by regulatory authorities before the release of a drug into the market have increased dramatically. In the discovery phase, each drug cost about $350 million.
- The Food and Drug Association processes I, II and III cost another $150 million. This brings the total to about $500 million for each drug put on the market for consumers.
- Due to these factors, the process of drug discovery is undergoing a complete overhaul to be cost-effective and to meet the supply and demand fundamentals.
Approaches For Drug Discovery
Evidence of the use of medicines and drugs can be found as far back in time as 3100 BC. The current scenario of the development of new drugs needs no emphasis in light of the current global situation of health and disease.
- For the majority of the time, drug discovery has been a trial-and-error process.
- Conventionally, the process of drug development has revolved around an almost blind screening approach, which was very time-consuming and laborious.
- The disadvantages of conventional drug discovery as well as the allure of a more deterministic approach to combat the disease have led to the concept of “Rational drug design”
in the 1960’s. A new understanding of the quantitative relationship between structure and biological activity ushered in the beginning of computer-aided drug design (CADD).
How To Design A Drug?
At the onset, it is important to know what features an “ideal” drug should have. The drug
- Nowadays, after knowing the detailed information of the target and lead molecule, a drug is designed with the help of computer tools.
- This can potentially save pharmaceutical companies, government, and academic laboratories alike from pursuing the “wrong” leads.
Structure-Based Drug Design
Structure-based drug design (SBDD) is considered one of the most innovative and powerful approaches in drug design. SBDD is an iterative approach. It requires a three-dimensional (3D) structure of the target protein, preferentially complexed with a ligand, where binding mode, affinity, and confirmation of a ligand binding can be discerned.
- Subsequently, various methods are used to design a high-affinity inhibitor either via virtual computer screening of large compound libraries or through the design and synthesis of novel ligands.
- Designed compounds are then tested in appropriate assays and the information is further used to guide the SBDD.
- Recent advances in computational methods for lead discovery include various commercially available software for de novo drug design, iterative design, selectivity discrimination, and estimation of ligand-binding affinities.
- SBDD and the emergence of structural genomics are paving the way to develop designer drugs. Two approaches to SBDD, the docking of known compounds into a target protein and de novo drug design have been merging as a single robust and powerful tool.
- In addition, the dynamics simulation of multiple copies of molecular building blocks in the presence of a receptor molecule is also a useful strategy for drug design, In the future, SBDD will merge with high throughput and, informatics technologies such as bioinformatics to design drugs with multiple homologous targets simultaneously.
- Dynamic combinatorial chemistry is a recently introduced supramolecular approach that uses self-assembly processes to generate libraries of chemical compounds. In contrast to the stepwise methodology of classical combinatorial techniques, dynamic combinatorial chemistry allows for the generation of libraries based on the continuous interconversion between the library constituents.
- Spontaneous assembly of the building blocks through reversible chemical reactions virtually encompasses all possible combinations, and allows the establishment of adaptive processes owing to the dynamic interchange of the library constituents.
- The addition of the target ligand or receptor creates a driving force that favors the formation of the best-binding constituent- a self-screening process that is capable, in principle, of accelerating the identification of lead compounds for drug discovery.
Drug Design Based On Bioinformatics Toots
The processes of designing a new drug using bioinformatics tools have opened a new area of research. However, computational techniques assist one in searching drug targets and designing drugs in silico, but it is time-consuming and expensive.
- Bioinformatics tools can provide information about potential targets that include nucleotide and protein sequencing information, homologs, mapping information, gene and protein expression data, function prediction, pathway information, disease associations, variants, structural information, and taxonomic distribution among others.
- This means that time, effort, and money can be saved in characterizing different targets. The field of. bioinformatics has become a major part of the drug discovery pipeline, playing a key role in validating drug targets.
- By integrating data from many interrelated yet heterogeneous resources, bioinformatics can help in our understanding of complex biological processes and help improve drug discovery.
Computer-Aided Drug Design
Computational tools have become increasingly important in drug discovery and design processes. Methods from computational chemistry are used routinely to study drug-receptor complexes in atomic detail and to calculate the properties of small-molecule drug candidates.
- Tools from information sciences and statistics are increasingly essential to organize and manage the huge chemical and biological activity databases that all pharmaceutical companies now possess and to make optimal use of these databases.
- In addition, the act of generating chemical derivatives is highly amenable to computerized automation. Libraries of derivative compounds are assembled by the application of targeted structure-based combinatorial chemistry from the analysis of active sites.
- Because of the combinatorial nature of this method, a large number of candidate structures may be possible. A computer can rapidly generate and predict the binding of all potential derivatives, creating a list of the best potential candidates. In essence, the computer filters all weak binding compounds, allowing the chemist to focus, synthesize, and test only the most promising ligands. Thus, using the CADD software to aid in the refinement of lead molecules is the most effective manner in which these tools can be employed.
- The use of computer modeling to refine structures has become standard practice in modern drug design. So the current role of computers in drug design lies in:
Storing And Retrieving Information:
- Structures determined experimentally by X-ray crystallography for biological targets (enzymes) and drug molecules
- Molecules and activities to test the effect of small structural changes on biological activity
Information About Toxicity And Its Relationship To Structure
Visualization Of Molecules:
- Similarities/differences between drugs and receptors
- Interaction between drugs and receptors
Calculations:
- Interaction strengths
- Motion (dynamics)
Challenges In Computer-Aided Drug Design
Highly intellectual professionals with interdisciplinary knowledge of various facets of science, most importantly, biology, chemistry, and computation are required for CADD and this is a major challenge for this field.
- In scientific computing, accuracy and processing time are always important. Thus, to make the calculations in a finite period, a plethora of assumptions, significant approximations, and numerous algorithmic shortcuts have to be used. This, in turn, greatly diminishes the calculated accuracy of any ligand-receptor interaction.
- This remains the most significant challenge in CADD. Another problem is the generation of a vast number of undesired chemical structures as there are a nearly infinite number of potential combinations of atoms and most of them are either chemically unstable, synthetically unfeasible, or have higher toxicity.
- Keeping in mind these shortcomings of CADD, improved generation of software with more user-friendly programs, superior and fast computational facilities, and creation of synthetic feasible and stable chemical compounds with refinement features have been developed in the last decade.
Drug Design Softwares
General Approach:
- The development of a new drug starts with the design of suitable candidate compounds, so-called “Ligands,” which are selected based on how the target protein recognizes these compounds and binds to them.
- “Ligbuild” is a powerful tool to build a legend just based on a protein structure in Brookhaven format. Performing experiments to know protein dynamics is expensive as well as time-consuming. The only alternative, computer simulation of the dynamics of the molecule (MD simulation), becoming increasingly important to identify which molecular properties are important and what are molecular interactions responsible for binding. Evaluation of binding agent is done by scoring approach.
- Score” is a tool to evaluate the binding affinity of protein-ligand complex with known 3D structure. Candidate molecules are further screened out on several criteria. Permeability across the biomembrane is an important characteristic.
- XLOGP can calculate logP (logarithm of the partition coefficient of a solute between octanol and water) of the common organic compounds, Furthermore, XLOGP can provide detailed hydrophobicity distribution information of the molecule.
- PLOGP can calculate logP values of peptides along with the Molecular Lipophilicity Potential (MLP) profile of a protein with a known structure. A database-based predictive system is also developed to assess the risk and toxicity of the chemicals in the early stage of drug design. The activity prediction studies based on the shape of the molecule include
- Fast and efficient clustering of molecules based on molecular shape
- Field-based similarity computation of molecular structure
- Flexible Quantitative Structure-Activity Relationships (QSAR) analysis of molecules based on shape cluster
- Comparative Molecular Field Analysis (CoMFA) has been widely used as a type of 3D QSAR method during the last 10 years.
Rational Programs Used: Drug design programs fall into one of three main categories: scanners, builders, or hybrids. Scanners-These types of programs are used for screening lead compounds. All database search programs fall into this category.
Rational Programs Used Strengths:
- Complete control of user on query specifications
- Established synthetic feasibility of compounds tested
- Rapid determination of potential binding ligands
- No scoring function is required
Rational Programs Used Weaknesses:
- The requirement for a wide database of structures
- Diversity of potential hits is limited as there is no recombination or derivatization of retrieved structures
Builders And Hybrids:
- These programs are mainly used for de novo generation of lead compounds. In these, the database contains fragments or chemical building blocks instead of complete compounds and requires the attachment point of the weak binding protein.
- It creates a population of derivatives with improved receptor complementarity by recombination or derivatization from fragments by making incremental changes iteratively.
Builders And Hybrids Strengths:
- No database of structures required
- Offers a vast number of potential derivative structures
- Creates truly novel ligands …
Builders And Hybrids Weaknesses:
- Questionable synthetic feasibility of compounds
- Generation of chemically unstable structures
- It depends mainly on the ability of the developer The software used
- Some of the frequently used software for drug design and their salient features are as follows:
Affinity:
- Automated, flexible docking
- Uses the energy of the ligand/receptor complex to automatically find the best binding modes of the ligand to the receptor (energy-driven method)
Auto Dock (Automated Docking Of Flexible Ligands To Receptors):
- It consists of three separate programs: AutoDock performs the docking of the ligand to a set of grids describing the target protein AutoGrid precalculates these grids AutoTors sets up which bonds will be treated as rotatable in the ligand
- Provide an automated procedure for predicting the interaction of ligands with biomolecular targets and help to narrow the conformational possibilities and in the identification of the most suitable structure
- Uses a Monte Carlo (MC) simulated annealing (SA) technique for configurational exploration with a rapid energy evaluation using grid-based molecular affinity potentials
- A powerful approach to the problem of docking a flexible substrate into the binding site of a static protein
- It has applications in X-ray crystallography, SBDD, lead optimization, virtual screening, combinatorial library design, protein-protein docking, and chemical mechanism studies
Combibuild:
- Structure-based drug design program created to aid the design of combinatorial libraries
- Screens library possible reactants on the computer, and predicts which ones will be the
most potent - Successfully applied to find nanomolar inhibitors of Cathepsin D
Dock Vision: A docking package created by scientists for scientists by including Monte Carlo, Genetic Algorithm, and database screening docking algorithms
Fred:
- Accurate and extremely fast, multi-conformer docking program
- Examines all possible poses within a protein active site, filtering for shape complementarities and optional pharmacophoric features before scoring with more conventional functions
Flexi Dock:
- Simple, flexible docking of ligands into binding sites on proteins
- Fast genetic algorithm for the generation of configurations
- Rigid, partially flexible, or fully flexible receptor side chains provide optimal control of ligand binding characteristics
- Conformationally flexible ligands
- Tunable energy evaluation function with special H-bond treatment
- Very fast run times
Flexx:
- A fast computer program for predicting protein-ligand interaction
- Two main applications: Complex prediction (create and rank a series of possible
protein-ligand complexes) Virtual screening (selecting a set of compounds for experimental testing) - Conformational flexibility of the ligand; rigid protein
- Placement algorithm based on the interactions occurring between fhe molecules (limited to low-energy structures)
- MIMUMBA torsion angle database used for the creation of conformers; interaction geometry database used to exactly describe intermolecular interaction patterns
- Boehm function (with minor adaptations necessary for docking) applied for scoring
Glide:
- High-throughput ligand-receptor docking for fast library screening
- Fast and accurate docking program
- Identifies the best binding mode through Monte Carlo sampling
- Provides an accurate scoring function for ranking binding affinities
- Can enrich the fraction of suitable lead candidates in a chemical database by predicting binding affinity rapidly and with a reasonable level of accuracy-will greatly enhance the probability of success in a drug discovery program
Gold:
- Calculates docking modes of small molecules into protein binding sites Based on genetic algorithm for protein-ligand docking
- Studies full ligand and partial protein flexibility
Predicts energy functions partly based on conformational and non-bonded contact information from the CSD - Choice of scoring functions: GoldScore, ChemScore, and User-defined score
- Has virtual library screening
Hint:
- Hydropathic Interactions
- Empirical molecular modeling system with new methods for de novo drug design and protein or nucleic acid structural analysis
- Translates the well-developed Medicinal Chemistry and QSAR formalism of LogP and hydrophobicity into a free energy interaction model for all bimolecular systems based on the experimental data from solvent partitioning Calculates 3D hydropathy fields and 3D hydropathic interaction maps
- Estimates LogP for modeled molecules or data files
Conclusions:
- The development of new drugs with potential therapeutic applications is one of the most complex and difficult processes in the pharmaceutical industry.
- Millions of dollars and man-hours are devoted to the discovery of new therapeutic agents. As the activity of a drug is the result of a multitude of factors such as bioavailability, toxicity, and metabolism, rational drug design has been a utopia for centuries.
- Very recently, impressive technological advances in areas such as structural characterization of biomacromolecules, computer sciences, and molecular biology have made rational drug design feasible. CADD is no longer merely a promising technique.
- It is a practical and realistic way of helping the medicinal chemist. On its own, it is unlikely to lead to pharmaceutical novelties but it has become a significant tool, an aid to thought, and a guide to synthesis. Still, drugs that are synthesized and tested by computational techniques can contribute a clear molecular rationale and above all provide a spur to the imagination.
Quantitative Structure-Activity Relationship: QSAR is building a mathematical model correlating a set of structural descriptors of a set of chemical compounds to their biological activity.
QSAR Introduction:
QSAR involves the derivation of a mathematical formula that relates the biological activities of a group of compounds to their measurable physicochemical parameters. These parameters have a major influence on the drug activity. The QSAR-derived equation takes the general form Biological activity = function (parameters)
Activity is expressed as log (1/c). C is the minimum concentration required to cause a defined biological response.
QSAR Parameters:
- The parameter is the measure of the potential contribution of its, group to a particular property of the parent drug.
- Various parameters used in QSAR studies are
- Lipophilic parameters: partition coefficient, n-substitution constant.
- Polarizability parameters: molar refractivity, paracord
- Electronic parameters: Hammetconstant, dipole moment.
- Steric parameters: Taft’s constant.
- Miscellaneous parameters: molecular weight, geometric parameters.
Lipophilic Parameters: Lipophilicity is the partitioning of the compound between an aqueous and non-aqueous phase.
Partition Coefficient:
P=[drug] in octanol/[drug] in water
- Typically over a small range of logP, for example, 1-4, a straight line is obtained.
Logl/C =0.75logP+2.30
- If the graph is extended to very high log P values, then get a parabolic curve
Logl/C=-kl (logP) 2+k 2logP+k3
- When P is small, dominated by log P term When P is large, log P squared dominates and so activity decreases
- substituent constant or hydrophobic substituent constants:
- The then-substituent constant is defined by Hanschand co-workers by the following equation.
Px= log Px-log PH
- A positive value indicates that the substituent has a higher lipophilicity than hydrogen and the drug favours the organic phase.
- A negative value indicates that the substituent has a lower lipophilicity than hydrogen and the drug favors the aqueous phase.
Electronic Parameters:
The Hammett Constant:
(σ); Sx= log (Kx/Kbenzoic)
- Equilibrium shifts Right and Kx – log Kbenzoic
- Since Sx= log Kx-log Kbenzoic, then s will be positive.
- Hammett constant takes into account both resonance and inductive effects; thus, the value depends on whether the substituent is para or meta-substituted Orthonot measured due to steric effects.
Steric Substitution Constant: It is a measure of the bulkiness of the group it represents and it affects the closeness of contact between the drug and receptor site. Much harder to quantify.
Steric Substitution Constant Examples Are:
- Taft’s steric factor (Es) (1956), an experimental value based on rate constants
- Molar refractivity (MR)- a measure of the volume occupied by an atom or group equation includes the MW, density, and the index of refraction
Ver loopsteric parameter-computer program uses bond angles, van derwaals radii, bond lengths
Hansch Analysis:
Proposed that drug action could be divided into 2 stages:
- Transport
- Binding Each of these stages depends upon the physical and chemical properties of the drug.
Log1/c=k1P=k2P2+k23s+k3Es+ks
- Look at the size and sign for each component of the equation. Values of 0.9 indicate the equation is not reliable Accuracy depends on using enough analogs, accuracy of data, and choice of parameters
- Applications: used to predict the activity of an as-yet unsynthesized analog.
Free Wilson Analysis:
This method is based on the assumption that the introduction of a particular substituent at a particular molecular position, always leads to a quantitatively similar effect on the biological potency of the whole molecules and is expressed by the equation.
Free Wilson Analysis Application:
- Easy to apply
- Simple method
- The substituent that cannot fulfill the principle of additivity can be recognized as effective when substituent constants are not available.
Topliss Method:
- This approach is completely non-mathematical and nonstatistical and does not need computerization of the data.
- A Toplissscheme is a flow diagram that in a series of steps directs the medicinal chemist to produce a series of analogs, some of which have greater activity than the lead used to start the 4 trees. There are two toplissschemes
- for the aromatic substituents
- for the aliphatic side chain substituents.
Topliss Method Applications: This method can be used if the synthetic route might be difficult and only a very few structures can be made in a limited time
Pharmacophore Modeling
Pharmacophore modeling is a successful yet very diverse subfield of computer-aided drug design. The concept of the pharmacophore has been widely applied to the rational design of novel drugs.
- In this paper, we review the computational implementation of this concept and its common usage in the drug discovery process.
- Pharmacophores can be used to represent and identify molecules on a 2D or 3D level by schematically depicting the key elements of molecular recognition. The most common application of pharmacophores is virtual screening, and different strategies are possible depending on the prior knowledge.
- However, the pharmacophore concept is also useful for ADME-tox modeling, side effects, and off-target prediction as well as target identification. Furthermore, pharmacophores are often combined with molecular docking simulations to improve virtual screening. We conclude this review by summarizing the new areas where significant progress may be expected through the application of pharmacophore modeling; these include protein-protein interaction inhibitors and protein design.
What Is A Pharmacophore?
- Historical perspective
- The original concept of the pharmacophore was developed by Paul Ehrlich during the late ISOOs.At that time, the understanding was that certain “chemical groups” or functions in a molecule were responsible for a biological effect, and molecules with similar effects had similar functions in common.
- The word pharmacophore was coined much later, by Schueler in his 1960 book Chemobiodynamics and Drug Design, and was defined as “a molecular framework that carries (phoros) the essential features responsible for a drug’s (pharmacon) biological activity.”
- The definition of a pharmacophore was therefore no longer concerned with “chemical groups” but “patterns of abstract features.”
- Since 1997, a pharmacophore has been defined by the International Union of Pure and Applied Chemistry as:
- A pharmacophore is the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response.
- The pharmacophore should be considered as the largest common denominator of the molecular interaction features shared by a set of active molecules. Thus a pharmacophore does not represent a real molecule or a set of chemical groups but is an abstract concept.
- Despite this clear definition, the term pharmacophore is often misused by many in medicinal chemistry to describe simple yet essential chemical functionalities in a molecule (such as guanidine or sulfonamides), or common’ chemical scaffolds (such as flavones or prostaglandins).
- Often the long definition is simplified to “A pharmacophore is the pattern of features of a molecule that is responsible for a biological effect,” which captures the essential notion that a pharmacophore is built from features rather than defined chemical groups.
Future Perspectives On Pharmacophore Modeling
Pharmacophore modeling has been around since the beginning of CADD and has evolved from a basic concept into a well-established CADD method with applications including similarity metrics, virtual screening, ligand optimization, scaffold hopping, target identification, and so on.
- Given the simplicity and versatility of the pharmacophore concept, it can be anticipated that further developments will be made in the future for different applications.
Fragment-Based Drug Design
Over the last two decades, fragment-based drug design has become a well-established method for the rational development of novel drugs. Rather than screening drug-like molecules (with molecular weights of around 500 Da), smaller molecules with a molecular weight of up to 350 Da (referred to as fragments) are being screened for affinity with a receptor using susceptible biophysical methods.
- Fragments showing some affinity for the target are grown into bigger and more potent compounds, and fragments binding to adjacent areas can be linked as well.
- Since the diversity of small molecule fragments can easily be sampled with a few hundred compounds, in silico screening methods are highly suitable for fragment-based design.
- CADD methods such as docking and pharmacophore modeling have therefore also been used to identify fragment-like compounds in silico before testing in vitro; subsequent fragment recombination can be used for the de novo design of inhibitors.
- In the first approach, the starting point is a single pharmacophore query that spans two (qr^^ more) sub-pockets in the receptor binding site. An additional pharmacophore feature is added that does not represent a molecular recognition feature but represents an atom in the fragments, where the two fragments of the different pockets may overlap and be linked.
- Then fragments are identified that fulfill the features present in a sub-pocket of the pharmacophore query, as well as on the linking feature.
- Then the compatibility of the fragment hits for the respective sub-pockets is evaluated in terms of the possibility of maintaining the correct conformation after linking the two fragments. Subsequently, the de novo-designed compounds can be synthesized and evaluated.
- In the following example, using a different yet similar strategy, Cavalluzzo et al designed a novel small molecule inhibitor binding to the LEDGF p75 protein, based on an inhibitory peptide.
- They used predefined amino acid side chain fragments taken from the inhibitory peptide and constructed a pharmacophore query to link the two predefined fragments with a third scaffold fragment that mimicked similar interactions as the peptide.
- All possible compounds were enumerated virtually, and for the compounds that were able to adopt a conformation similar to the pharmacophore query after linking all fragments, the chemical synthesizability was assessed. Following synthesis, the inhibitory potency of the compound was found to be 30 pM IC50 compared to 7.4 pM IC50 for the most potent inhibitory peptide.
- Even when active fragments have been identified using the classical in vitro methods, computational pharmacophore methods can be applied to identify novel derivatives.
- For example, pharmacophore fingerprint-based similarity searching and the generation of 3D pharmacophore queries are suitable means to identify bigger and more potent molecules from small molecule libraries.
Protein-Protein Interaction (PPI) Inhibition:
Although once thought to be undruggable, “high-hanging fruits on the drug discovery tree/’ PPIs have drawn a great deal of attention in recent years. The undruggable image has disappeared, and several small molecule inhibitors of PPIs (SMPPII) have been reported. Most of the early inhibitors originate from HTS.
- Structural analysis of proteins in PPI complexes and inhibitor complexes shows that the interactions at the PPI interface are being mimicked by the ligand.
- SMPPII are found to copy the natural interaction not only in terms of shape and chemistry but even at the electrostatic potential level. This mimicry suggests that the pharmacophore queries created from PPI complex structures can be used to identify SMPPII via virtual screening. Different methods can be employed to map the pharmacophore features onto the amino acids present at the PPI interface.
- Several SMPPII discoveries have been achieved, thanks to pharmacophore searches using manually created search features, a consensus of interactions at the PPI interface, or using automated methods, or by identification of the key interactions using molecular interaction field analysis.
- PPIs are especially promising targets for controlling inappropriate signaling, as found in diseases such as cancer. The usefulness of pharmacophore modeling to create queries encoding the key interactions at the PPI interface will probably strongly stimulate the discovery of novel SMPPII using pharmacophores, both as a stand-alone virtual screening tool and incorporated into pipelines with other methods
A Potential Role In Protein Design?
Although pharmacophore modeling originated as a drug design concept and, as indicated earlier, is nowadays a key element of CADD, pharmacophore modeling shows promise in the currently burgeoning field of computational protein design.
- Rather than designing drugs for a given protein target, computational protein design aims to derive an amino acid sequence that will fold into a given structure with a desired function.
- In many cases, this may involve protein-small molecule ligand interactions, and for these, it can easily be imagined that pharmacophores may be used simply by reversing the process of small molecule drug design for a known protein structure.
- First of all, suitable protein templates (enzymes or otherwise) should be identified for the protein redesign process. The ligand of interest could serve as a query to try to identify possible binding proteins, which can then later be redesigned to give optimum complementarity to the ligand.
- Second, during the virtual protein design process, often multiple rotamers of different amino acids are sampled to identify the most desirable ones.
- Similar to ligand fitting with a pharmacophore query, the protein side chains can be fitted to features describing the complementary interactions required at the protein-ligand interface.
A Potential Role In Protein Design Importance
- The pharmacophore concept was first put forward as a useful picture of drug interactions almost a century ago, and with the rise in computational power over the last few decades, has become a well-established CADD method with numerous different applications in drug discovery.
- Depending on the prior knowledge of the system, pharmacophores can be used to identify derivatives of compounds, change the scaffold to new compounds with a similar target, virtual screen for novel inhibitors, profile compounds for ADME-tox, investigate possible off-targets, or just complement other molecular methods.
- While there are limitations to the pharmacophore concept, multiple remedies are available at any time to counter them. Given this versatility, it is expected that pharmacophore modeling will maintain a dominant role in CADD for the foreseeable future, and any medicinal chemist should be aware of its benefits and possibilities.
Drug Design Multiple Choice Question And Answers
Question 1. The full form of CADD is.
- Central Approach to Drug Design
- Command Attribute for Drug design
- Computer-Aided Drug Design
- None
Answer: 4. None
Question 2. What is the full form of SBDD?
- Software-Based Drug Design
- Structure-Based Drug Design
- Sample-Based Drug Design
- None
Answer: 3. Sample-Based Drug Design
Drug Design Short Question And Answers
Question 1. What is Drug design?
Answer:
Drug Design: Drug design is the inventive process of finding new medications based on the knowledge of a biological target.
- In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the molecular target with which they interact and bind.
- Drug design frequently but not necessarily relies on computer modeling techniques and bioinformatics approaches in the big data era.
Question .2 Write the Ideal characteristics of a Drug.
Answer:
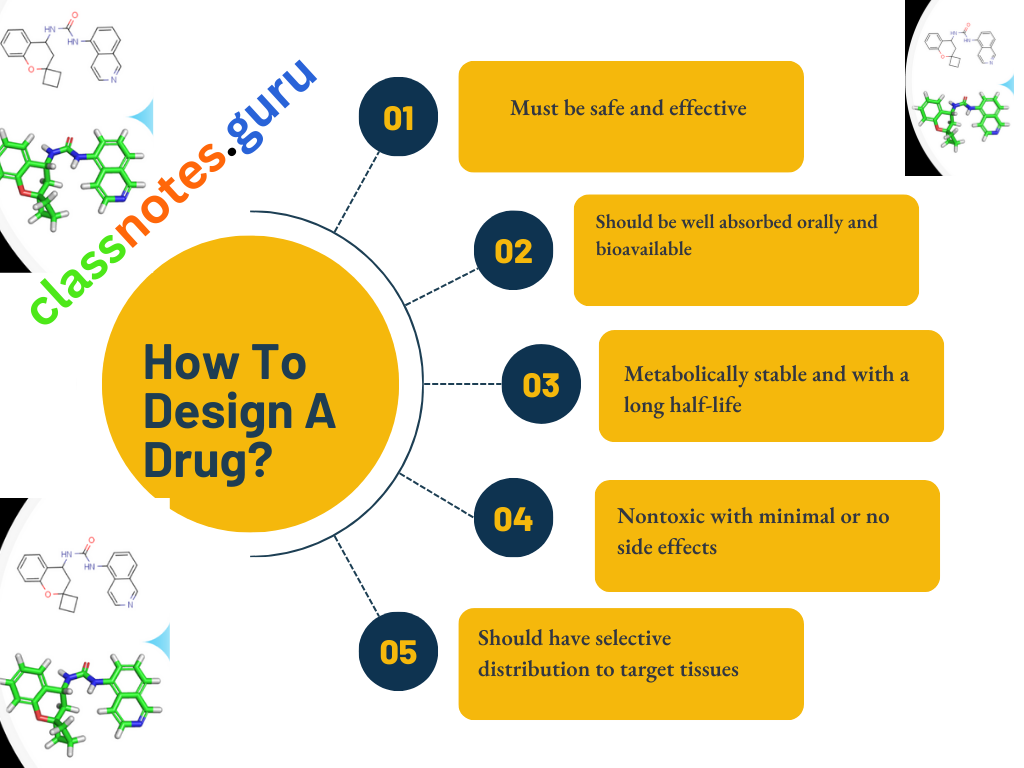
The Ideal Characteristics Of A Drug
- The ideal characteristics of a drug are.
- Must be safe and effective
- Should be well absorbed orally and bioavailable
- Metabolically stable and with a long half-life
- Nontoxic with minimal or no side effects
- Should have selective distribution to target tissues
Question .3 What is QSAR?
Answer:
QSAR: QSAR involves the derivation of a mathematical formula that relates the biological activities of a group of compounds to their measurable physicochemical parameters. These parameters have a major influence on the drug’s activity.