Disorders Of Platelets Important Notes
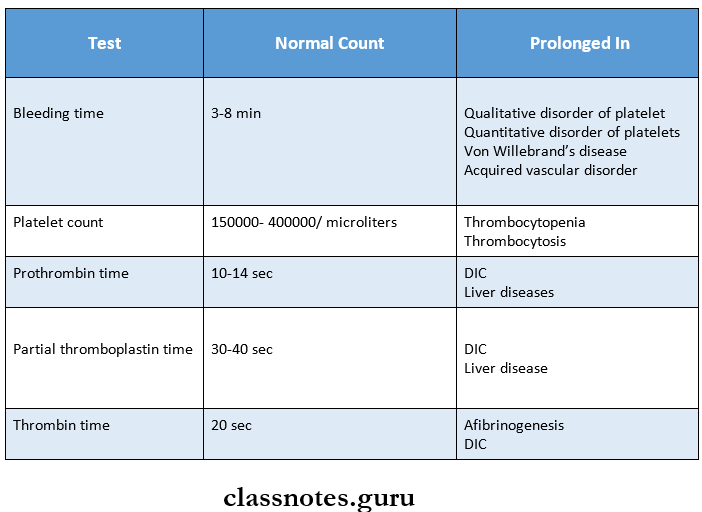
1. Partial thromboplastin time
- Used to measure the intrinsic factors as well as factors common to both intrinsic and extrinsic pathways
- The normal range is – 30-40 sec
- Causes of prolonged partial thromboplastin time are
- Parenteral administration of heparin
- DIC
- Liver disease
- Circulating anticoagulants
2. Thrombin time
- It is a semi-quantitative test for fibrinogen deficiency
- Normal range – 20-30 sec
- Common causes of prolonged thrombin time are
- Presence of heparin
- DIC
3. Bleeding time
- Normal range – 3-8 min
- Prolonged in
- Thrombocytopenia
- Von Willebrand disease
- Vascular abnormalities
- Disorders of platelet functions
4. Clotting time
- Normal range – 4-9 min
- Prolonged in
- Hemophilia
- Vitamin K deficiency
- Liver diseases
- Anticoagulant administration
5. Von Willebrand disease
- It occurs due to qualitative defect in Von Willebrand factor in blood
- It is characterized by
- Normal platelet count
- Prolonged bleeding time
- Defective platelet aggregation
- Reduced factor 8 activity
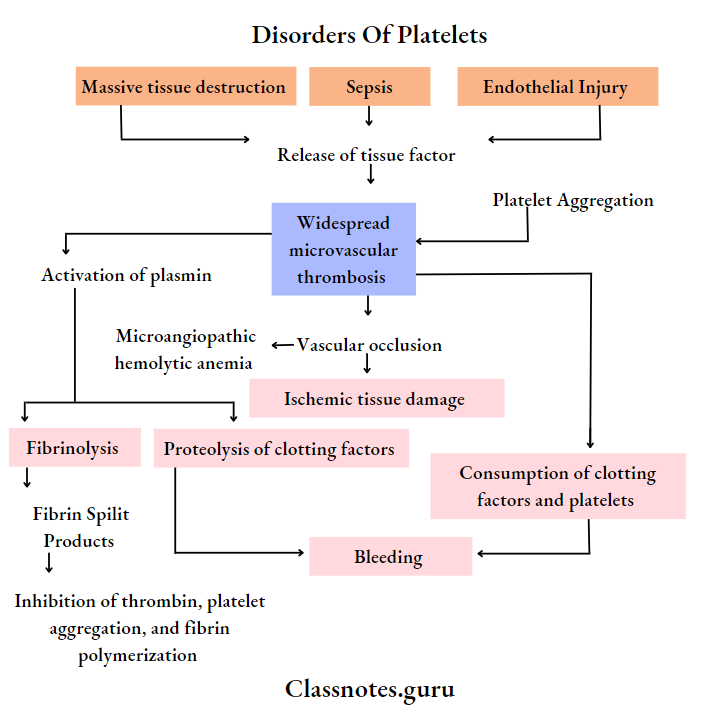
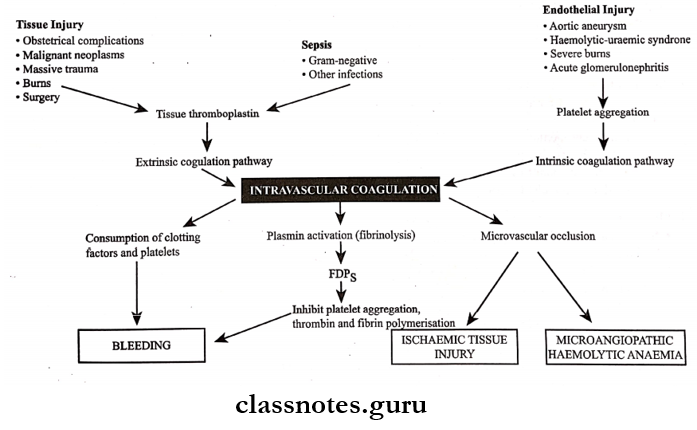
6. DIC (Disseminated Intravascular Coagulation)
- It is characterized by systemic activation of the blood coagulation system
- Results in the generation and deposition of fibrin leading to Microvascular thrombi in various organs
- Severe bleeding complications occur
- It is seen in promyelocytic leukaemia
Disorders Of Platelets Long Essays
Question 1. What is disseminated intravascular coagulation? Describe its etiopathogenesis.
Answer:
Disseminated Intravascular Coagulation: Disseminated intravascular coagulation is a complex thrombo-hemorrhagic disorder occurring as a secondary complication in some systemic diseases
Disseminated Intravascular Coagulation Etiopathogenesis: Pathogenesis of DIC includes the following events
Read And Learn More: Pathology Question And Answers
1. Activation of coagulation: Etiological factors like massive tissue injury, presence of infections, and endothelial damage cause activation of coagulation by the release of tissue factor
2. Thrombotic phase:
- Endothelial damage from various thrombogenic stimuli causes:
- Generalized platelet aggregation
- Platelet adhesion
- Deposition of small thrombi and emboli throughout microvasculature
3. Consumption phase: Consumption of coagulation factors and platelets occurs
4. Secondary fibrinolysis:
- The fibrinolytic system is secondary activated
- This causes the breakdown of fibrin resulting in the formation of FDPs in circulation
Disorders Of Platelets Short Essays
Question 1. Bleeding time
Answer:
- Bleeding time (BT] is defined as the time lapse between skin puncture and the arrest of bleeding.
- BT is the time from the onset of bleeding to the stoppage of bleeding. The bleeding stops due to the formation of a temporary haemostatic plug.
- Procedure: Under aseptic conditions, give a deep finger prick and note the time. Then remove the blood drop every 15 s with a clotting paper (along the edges] at a different spot till the bleeding stops. Count the number of spots and calculate BT as follows:
- BT = Initial time + (number of spots – 1] x 15 s
Bleeding Time Indications:
- It is a useful screening test in patients with a history of prolonged bleeding.
- In patients with bleeding disorders before any surgical procedures.
Bleeding Time Interpretation: An abnormal BT is usually the result of
- Abnormalities in the structure or ability of capillary blood vessels to contract or
- Abnormalities in the number or functional integrity of the platelets.
Question 2. Thrombocytopenia
Answer: Thrombocytopenia is defined as a reduction in the peripheral blood platelet count below the lower limit of normal i. e., below 1,50,000/microlitre
- Platelet counts in the range of 20,000 – 50,000 cells/microlitre are associated with an increased risk of post-traumatic bleeding.
- Spontaneous bleeding is evident when the count falls below 20,000 cells/microlitre
Thrombocytopenia Causes:
1. Impaired platelet production:
- Generalised bone marrow failure.
- Aplastic anaemia, leukaemia, megaloblastic anaemia, marrow infiltration.
- Selective suppression of platelet production.
- Drugs (Anticancer, cytotoxic, alcohol], Infection (HIV, measles].
2. Accelerated platelet destruction:
- Immunologic destruction.
- Autoimmune – ITP, SLE
- Drug associated – Heparin, sulfa compounds.
- Infections – HIV, cytomegalovirus infections.
- Non – immunologic destruction,
- DIC
- Gaint haemangiomas
- TTP
3. Splenic Sequestration: Splenomegaly.
4. Dilutional loss: Massive transfusion of old stored blood to bleeding patients.
Thrombocytopenia Clinical features:
- Usual manifestations are petechial haemorrhage, purpura, easy bruising, epistaxis, mucosal bleeding such as menorrhagia in women, nasal bleeding, bleeding from gums, and haematuria.
- Intracranial haemorrhage is rare.
- Splenomegaly and hepatomegaly may also occur.
Thrombocytopenia Investigations:
- Low platelet counts,
- Prolonged bleeding times,
- Normal coagulation profile
- Abnormal long clot retraction time.
Thrombocytopenia Treatment:
- Platelet transfusion.
- Treatment of the underlying cause.
Question 3. Hemophilia
Answer:
- Haemophilia is an X-linked recessive disorder characterised by the deficiency of factor 8 (Haemophilia A] classic haemophilia]
- Whereas inherited deficiency of factor 9 (Christmas factor/plasma thromboplastin component] produces Christmas disease/haemophilia B (Christmas disease].
Hemophilia A: Pathogenesis: It is caused by quantitative reduction of factor 8 in 90% of cases while 10% have normal/increased levels of factor 8 but reduced activity.
Hemophilia A Clinical features:
- In severe cases -Bleeding is spontaneous.
- In mild disease – Bleeding is rarely spontaneous
- Bleeding occurs mainly in joints (Haemarthrosis), muscles (haematoma), and viscera/in retroperitoneum but can involve any organ system.
- Spontaneous intracranial haemorrhage and oro-pharyngeal bleeding are rare, but when they occur they are the most feared complications.
Hemophilia A Treatment: Factor 8 replacement therapy, consisting of factor 8 concentrates/plasma cryoprecipitates.
Haemophilia B:
- Haemophilia B is rarer than haemophilia A.
- Inheritance patterns and clinical features of factor 9 are indistinguishable from those of classic haemophilia but accurate laboratory diagnosis is critical since haemophilia B requires treatment with different plasma fractions.
Hemophilia B: Treatment: infusion of either fresh frozen plasma/plasma enriched with factor 9
Question 4. Von Willebrand’s disease
Answer:
Von Willebrand’s Disease: This is the most common hereditary coagulation disorder occurring due to qualitative/quantitative defects in Von Willebrand’s factor.
- Von Willebrand factor is a multimeric plasma glycoprotein synthesized by megakaryocytes and endothelial cells. It serves two major functions as follows.
- Acts as a carrier protein for factor 8.
- Helps in the adhesion of platelets to subendothelial collagen.
Von Willebrand’s Disease Clinical features:
- Spontaneous bleeding from mucous membranes and excessive bleeding from wounds.
- Gastrointestinal bleeding.
- Epistaxis, menorrhagia, and superficial bruises are present
Von Willebrand’s Disease Lab investigations:
- Prolonged bleeding time,
- Normal platelets count,
- Bleeding time increased,
- Reduced vWF levels, factor 8 activity reduced.
Von Willebrand’s Disease Treatment:
- Factor 8 and vWF concentrate transfusion,
- The bleeding episodes can be managed by giving vasopressin which increases the VWF levels.
- Persistent bleeding is treated with factor 8 concentrate.
- Cryoprecipitate transfusion
- Antifibrinolytic agent
- Example: Tanexamie acid is useful in conjunctive therapy during dental procedures.
Question 5. Pancytopenia
Answer: Pancytopenia means the occurrence of anaemia, leukopenia and thrombocytopenia together.
The causes of pancytopenia are as follows:
1. Aplastic anaemia
2. Pancytopenia with normal or increased marrow cellularity.
- Myelodysplastic syndromes
- Hypersplenism
- megaloblastic anaemia
3. Paroxysmal nocturnal haemoglobinuria
4. Bone marrow infiltrations.
- Haematologic malignancies (leukaemias, lympho-mas, myelomas)
- Nonhaematologic metastatic malignancies
- Storage diseases
- Osteopetrosis.
Question 6. Idiopathic Thrombocytopenic Purpura.
Answer: It is characterized by the immunologic destruction of platelets and normal or increased megakaryocytes in bone marrow
Idiopathic Thrombocytopenic Purpura Types:
- Acute
- Chronic
Idiopathic Thrombocytopenic Purpura Clinical Features:
- Usual manifestations
- Petechial haemorrhage
- Purpura
- Easy bruising
- Epistaxis
- Mucosal bleeding such as menorrhagia in women
- Nasal bleeding
- Bleeding from gums
- Hematuria
- Intracranial haemorrhage is rare
- Splenomegaly and hepatomegaly may also occur
Idiopathic Thrombocytopenic Purpura Lab Diagnosis:
- Diagnosis can be made from the following findings
- Thrombocytopenia
- Microangiopathic haemolytic anaemia
- Leucocytosis
- Bone marrow aspiration shows increased megakaryocytes
- Examination of biopsy shows typical microthrombi in arterioles, capillaries and venules
Disorders Of Platelets Short Question And Answers
Question 1. Prothrombin time
Answer:
- Prothrombin measures the extrinsic system factor 7 as well as factors in the common pathway
- In it, tissue thromboplastin and calcium are added to the test
Normal Time: 10-14 seconds
Prolonged In:
- Administration of oral Anticoagulants
- Liver disease
- Vitamin K deficiency
- Disseminated intravascular coagulation
Question 2. Screening tests for bleeding disorders
Answer: