
Class 11 Chemistry Organic Chemistry Basic Principles And Techniques Organic Chemistry Purification And Analysis Of Organic Compounds Introduction
The presence of impurities even in very small amounts may sometimes result in deviation of some properties of organic compounds to a marked degree. Therefore, to characterise an organic compound thoroughly, it is essential to obtain it in the purest form. Again, an organic compound must be in a certain state of purity before it can be analysed qualitatively and quantitatively to arrive at its correct molecular formula. The organic compounds whether isolated from a natural prepared in the laboratory are mostly impure. These are generally contaminated with some other substances. A large number of methods are available for the purification of organic compounds.
Read and Learn More CBSE Class 11 Chemistry Notes
Different Methods For The Purification Of Organic Compounds
Some of the important methods which are commonly employed for the purification of organic compounds are as follows:
- Crystallisation
- Sublimation,
- Distillation,
- Extraction and
- Chromatography.
1. Crystallisation
Crystallisation Definition:
Crystals are the purest form of a compound having definite geometrical shapes and the process by which an impure compound is converted into its crystals is known as crystallisation.
This is one of the most commonly used methods for the purification of solid organic compounds. It is based on the difference in solubilities ofthe compound and the impurities in a suitable solvent.
A solvent is said to be the most suitable one which fulfils the conditions such as:
- The organic solid must dissolve in the solvent on heating and must crystallise out on cooling
- The solvent must not react chemically with the organic compound, and
- The impurities should not be normally dissolved in the solvent or if they dissolve, they should be soluble to such an extent that they remain in the solution, i.e., in the mother liquor.
The various solvents which are commonly used for crystallisation are water, alcohol, ether, chloroform, carbon tetrachloride, benzene, acetone, petroleum ether etc.
Class 11 Chemistry Purification and Analysis of Organic Compounds Notes
Crystallisation Procedure:
- A certain amount of an impure organic compound is added to a minimum amount of a suitable solvent and the mixture is then heated to get a hotsaturated solution ofthe compound. The hot solution is then filtered to remove the insoluble impurities, if present.
- The clear solution is then allowed to cool down undisturbed when the solid organic compound separates in the form of fine crystals. The crystals do not separate even after a long time, the inner surface of the vessel is scratched with the round end of a glass rod to facilitate crystallisation.
- The addition of a few crystals of the pure compound to the solution may also hasten the crystallisation process. The process of inducing crystallisation by adding a few crystals of the pure compound into its saturated solution is called seeding.
- The crystallised compound is then filtered as usual. The crystals on the filter paper are washed with a small amount of solvent to remove the impurities.
- The compound is then pressed in between the folds of filter paper to remove water as far as practicable. It is then dried in an esteem or air oven and finally in a vacuum desiccator.
Fractional crystallisation:
This method is used for the separation of a mixture of two (or more) compounds which have unequal solubilities in a particular solvent.
Fractional crystallisation Procedure:
- A saturated solution of the mixture of compounds is prepared in a suitable solvent by applying heat and the hot solution is then allowed to cool when the less soluble component crystallises out earlier than the more soluble component.
- The crystals are separated by filtration.
- The mother liquor is then concentrated and the hot solution is allowed to cool when the crystals of the more soluble second component are obtained.
- By repeating the process, all the components of the mixture are separated.
- It thus follows that fractional crystallisation is the process of separation of different components of a mixture by repeated crystallisation.
2. Sublimation
Sublimation Definition:
The Process of conversion of a solid into the gaseous state on heating without passing through the intervening liquid state and vice versa on cooling is called sublimation.
- Only those substances, whose vapour pressures become equal to the atmospheric pressure much before their respective melting points, undergo ready sublimation when heated
- This process is very useful for the separation of volatile solids which sublime on heating from the non-volatile impurities.
Sublimation Procedure:
- The impure sample is taken in a china dish covered with perforated filter paper (or porcelain plate).
- An inverted funnel is placed over the dish and its stem is plugged with cotton.
- The dish is then heated gently when vapours of the volatile substance pass through perforations of the filter paper and condense on the cooler walls of the funnel leaving behind non-volatile impurities in the dish
Sublimation Applications:
Benzoic acid, camphor, naphthalene, anthracene, iodine etc. are purified by this method. In the case of other compounds like indigo which are very susceptible to thermal decomposition, sublimation is done under reduced pressure
Purification of Organic Compounds Class 11 Chemistry Notes

3. Distillation
Distillation Definition:
The process of conversion of a liquid into its vapours by heating followed by condensation of vapours thus produced by cooling is called distillation
- The process of simple distillation is commonly used for the purification of liquids which do not undergo decomposition, on boiling Le., which are sufficiently stable at their boiling points and which contain non-volatile impurities.
- Organic liquids such as benzene, ethanol, acetone, chloroform, carbon tetrachloride, toluene etc. can be purified by the process of simple distillation
Distillation Procedure:
- The impure organic liquid is taken in a distillation flask which is fitted with a water condenser and a thermometer.
- A receiver is attached to the lower end of the condenser.
- One or two pieces of unglazed porcelain or glass beads are added to prevent bumping of the liquid during distillation.
- The flack is then heated in a water bath or a sand bath (in the case of volatile and inflammable liquid) or directly (in the case of liquids having high boiling points) when the temperature rises gradually.
- The liquid starts boiling when its vapour pressure becomes equal to the atmospheric pressure.
- The vapours then pass through the water condenser and condense to form the liquid which is collected in the receiver
- The non-volatile impurities are left behind in the distillation flask
Distillation:
Analysis of Organic Compounds Class 11 Chemistry Notes

1. Fractional distillation:
Fractional distillation Definition:
The”distillation process in which a mixture of two or more miscible liquids having boiling points close to each other are separated is called fractional distillation
If the boiling points ofthe two liquids of a mixture are very close to each other, i.e., differ only by 10- 20K, their separation cannot be achieved by a simple distillation method. In such cases, the separation can be achieved by fractional distillation which involves repeated distillation and condensations by using a fractionating column
Fractional distillation Procedure:
- The apparatus used is the same as in the simple distillation process except for a fractionating column.
- When the mixture is heated, the temperature rises slowly and the mature starts boiling. The formed mainly consists of the more volatile liquid with a little of the less volatile liquid.
- As these vapours travel up in the fractionating column, the vapours ofthe less volatile liquid condense more readily than those of the more volatile liquid.
- Therefore, vapours rising become rich in the vapours of more volatile liquid and the liquid flowing down becomes rich in less volatile liquid.
- This process is repeated throughout the length of the fractionating column.
- As a consequence, the vapours which escape from the top of the column into the condenser consist of almost the more volatile liquid.
- Thus, the distillate received contains the more volatile component almost in pure form whereas the liquid left behind in the flask is very rich in the less volatile component.
- This process may sometimes be repeated to achieve complete separation of liquids.

Fractional distillation Applications:
One of the most important applications of fractional distillation is to separate crude petroleum into various fractions like gasoline, kerosene oil, diesel oil etc. 0 Fractional distillation is also used to separate methanol (b.p. 338 K) and acetone (b.p. 329 K) from pyroligeneous acid obtained by destructive distillation of wood.
2. Distillation under reduced pressure (Vacuum distillation):
The distillation process which involves the purification of high boiling liquids (which decompose at or below their boiling points) by reducing the pressure over the liquid surface is called vacuum distillation.
Some liquids which tend to decompose at or below their boiling points cannot be purified by ordinary distillation. Such liquids can be purified by distillation under reduced pressure. A liquid boils when its vapour pressure becomes equal to the external pressure.
Therefore if the pressure acting on It is reduced, the liquid boils at a lower temperature and so, its decomposition does not occur. Glycerol, for example, decomposes at its boiling point (563 K). However, if the external pressure is reduced to 12 mm, it boils at 453 K without decomposition. Some other compounds such as phenylhydrazine, diethyl malonate, ethyl acetoacetate etc. are also purified by this method.
Vacuum distillation Procedure:
- The distillation is carried out in a two-necked flask called Claisen’s flask.”A capillary tube is fitted into one neck ofthe flask and is kept immersed in the liquid to be distilled.
- The other neck of the flask is fitted with a thermometer. The side tube of this neck is connected to a condenser carrying a receiver at the other end.
- The receiver is connected to a vacuum pump and a manometer.
- The flask is usually heated in a sand or oil bath. To prevent bumping, a steady flow of air is maintained by the capillary tube with the help ofthe screw-type cock attached to it The desired pressure is maintained by using the vacuum pump

3. Steam distillation:
Steam distillation is applied for the separation and purification of those organic compounds which
- Are insoluble in water
- Are steam volatile,
- Possess a vapour pressure of 10-15 mm Hg at 100°C and
- Contain non-volatile organic or inorganic impurities.
Steam distillation Principle:
In steam distillation, the liquid boils when the sum of the vapour pressures of the organic liquid (pt) and that of water (p1) becomes equal to the atmospheric pressure (p), i.e.,
p = p1 + p2. Since pt is lower than p, the organic liquid boils at a temperature lower than its normal boiling point and hence its decomposition can be avoided. Thus, the principle of this method is similar to that of distillation under reduced pressure.
NCERT Class 11 Chemistry Purification of Organic Compounds
Steam distillation Procedure:
- The impure organic liquid is taken in a roundbottom flask. Steam from a steam generator is passed into the flask which is gently heated.
- The mixture starts boiling when the combined vapour pressure becomes equal to the atmospheric pressure
- At this temperature, the vapours of the liquid with the steam escape from the flask and after getting condensed it is collected in the receiver.
- The distillate contains the desired organic liquid and water which can easily be separated by using a separating funnel

4. Differential extraction
Differential extraction Method:
The method of separation of an organic compound from its aqueous solution by shaking with a suitable solvent is called differential extraction
A solid or liquid organic compound can be recovered from its aqueous solution by shaking the solution in a separating funnel with a suitable organic solvent which is insoluble in water but in which the organic compound is highly soluble. Some commonly employed solvents for extraction are ether, benzene, chloroform, carbon tetrachloride etc.
Differential extraction Procedure:
- The aqueous solution ofthe organic compound is mixed with a small quantity of the organic solvent in a separating funnel
- The funnel is stopped and its contents are shaken vigorously when the organic solvent dissolves out of the organic compound.
- The separating funnel is then allowed to stand for some time when the solvent and water form two separate layers.
- The lower aqueous layer (when the organic solvent used is ether or benzene) is run out by opening the tap of the funnel and the organic layer is collected.
- The whole process is repeated to remove the organic compound completely from the aqueous solution.
- The organic compound is finally recovered from the organic solvent by distilling off the latter.

5. Chromatography
Chromatography Definition:
The technique used for the separation of the components of a mixture in which the separation is achieved by the differential movement of individual components through a stationary phase under the influence of a mobile phase is called chromatography.
Chromatography is the most useful and modern technique extensively used for the separation of mixtures into their components, to purify the compounds and also to test the purity of compounds.
This method was first invented by M. Tswett, a Russian botanist in 1906 for the separation of coloured substances into individual components. The word ‘Chromatography’ was originally derived from the Greek word chroma means colour and graphy means writing.
Types of chromatography:
Depending upon the nature of the stationary phase (either a solid or a tightly held liquid on a solid support) and the mobile phase (either a liquid or a gas),
The various types of chromatographic techniques commonly used are:
- Column or Adsorption chromatography,
- Thin Layer Chromatography (TLC),
- High-performance liquid Chromatography (HPLC),
- Gas Liquid Chromatography (GLC),
Paper or partition Chromatography. In the first three cases, the mobile phases are liquid and the stationary phases are solid.
In the fourth case, the mobile phase is gas while the stationary phase is liquid and in the fifth case, both the mobile and stationary phases are liquid.
The chromatographic separation is based on the principle that the components of the mixture present in the moving phase move at different rates through the stationary phase and thus get separated. Now, depending on the basic principle, chromatography can be divided into two categories:
Absorption chromatography and Partition chromatography.
NCERT Class 11 Chemistry Purification of Organic Compounds
Adsorption chromatography Principle:
This category of chromatography is based upon the differential adsorption of the various components of a mixture on a suitable adsorbent such as alumina, cellulose, silica gel, magnesium oxide etc. Since some compounds undergo adsorption better than others, they travel through the column at different rates and thus get separated.
Adsorption chromatography is of two types:
- Column chromatography and
- Thin Layer Chromatography (TLC).
1. Column chromatography
It is the simplest of all the chromatographic techniques and is extensively used.
Chromatography Procedure:
- A plug of cotton or glass wool is placed at the bottom of a clean and dry glass column and it is then covered with a layer of acid-washed sand.
- A suitable adsorbent such as alumina, silica gel, magnesium oxide, starch etc. is made into a slurry with a non-polar solvent such as hexane or petroleum ether and the slurry is then added to the column gradually and carefully so that no air bubble is entraped in the column.
- The excess of the solvent above the adsorbent is removed by opening the stop-cock. This constitutes the stationary phase.
- The mixture of compounds (say A, B and C) to be separated is dissolved in a minimum volume of a suitable highly polar solvent in which it is readily soluble. It is then added to the top of the column with the help of a dropper or a microsyringe and allowed to pass slowly through it (if the mixture is liquid, it is added as such).
- As the solution travels down, the different components of the mixture get adsorbed to different extents depending upon their polarity (say, A>B>C) and form a narrowband which is quite close to the top of the column. This band or zone is called a chromatogram.
- A suitable solvent called eluent is then made to run through the column. The polarity of the solvent is gradually increased to elute the adsorbed materials. The eluent acts as the mobile phase.
- As the solvent moves down the column, the components A, B and C present in the chromatogram begin to separate. The eluent dissolves out the different components selectively.
- The component which is strongly adsorbed on the stationary phase moves slowly down the column, whereas the component that is weakly adsorbed moves at a faster rate. Therefore, the three components [A, B and C) form three bands at different places in the column.
- As the addition of eluent is continued, the adsorbed components present in the bands are dissolved by the solvent and are then collected in the form of different fractions in separate conical flasks.
- The eluent from each flask is then removed by evaporation or distillation to get the various components in pure form.
- The process of separation of different components of the mixture from the adsorbent and their recovery with the help of a suitable solvent is called elution.

2. Thin Layer Chromatography (TLC):
It is another type of adsorption chromatography which involves the separation of the components of a mixture over a thin layer of adsorbent. This technique is particularly useful in rapid analysis ofthe purity of samples.
Chromatography Procedure:
- A thin layer (0.2 mm thick) of adsorbent such as silica gel or alumina is spread over a plastic or glass plate of suitable size (5 cm × 20 cm).
- This thin layer of adsorbent acts as the stationary phase. This plate is called a thin-layer chromatography plate or TLC plate or chromatophore.
- Two pencil lines are drawn across the width of the plate at distances about 1 cm from each end. The lower line is called the base line or starting line and the upper line is called the finish line or solvent front.
- A drop of the solution of the mixture to be separated is placed on the starting line with the help of a capillary.
- The plate is then dried and placed in a vertical position in a jar called a developing chamber containing a suitable solvent or a mixture of solvents. It acts as the mobile phase. The height of the solvent in the jar should be such that its upper surface does not touch the sample spot.
- The chamber is then closed and kept undisturbed for half an hour
- As the solvent slowly rises by the capillary action, the components of the mixture also move up along the plate to different distances depending upon their degree of adsorption and thus separation takes place.
- When the solvent front reaches the finish line, the plate is removed from the jar and then dried. The spots of coloured components, due to their original colour, is visible on the TLC plate.
The spots of the colourless components which are invisible to the eye can be detected:
- By placing the plate under a UV lamp because certain organic compounds produce a fluorescence effect in UV light
- By placing the plate in a covered jar containing a few crystals of iodine because certain organic compounds which absorb iodine turn brown and
- By spraying the plate with the solution of a suitable chemical reagent
- For example – Ninhydrin in case of amino acids; 2,4-dinitrophenylhydrazine in case of carbonyl compounds
- Different components developed on the TLC plate are identified through their retention factors (or retardation factor), i.e., R j values.
It may be defined as:
⇒ \(R_f=\frac{\text { Distance travelled by the compound from the baseline }}{\text { Distance travelled by the solvent from the baseline }}\)
- If the two components of the mixture, for example, are A and B, then according to the given their R2 values will be a/l and b/l respectively.
- The two compounds can be identified by comparing these Rf values with the Rf values of pure compounds. Since the solvent front on the TLC plate always moves faster than the compounds, Rf values are always less than 1.
- Each spot is finally eluted separately with suitable solvents and collected

Partition chromatography
Unlike adsorption chromatography (column chromatography or TLC) which represents solid-liquid chromatography, partition chromatography is a liquid-liquid chromatography, i.e., both the stationary phase and mobile phase are liquids.
Partition chromatography Principle:
Partition chromatography is based on continuous differential partitioning (distribution) of components of a mixture between the stationary and the mobile phases Paper chromatography is a common example of partition chromatography.
In paper chromatography, a special type of paper known as chromatographic paper is used. Although the paper is made up of cellulose, the stationary phase in paper chromatography is not the cellulose but water which is adsorbed or chemically bound to cellulose. The mobile phase is usually a mixture of two or three liquids with water as one ofthe components.
Organic Compound Purification Methods Class 11 Notes
Partition chromatography Procedure:
- A suitable strip of chromatographic paper (20cm × 5cm, Whatman filler paper) is taken and a starting line (baseline) is drawn across the width of the paper at about 1 to 2 cm from the bottom.
- The mixture to be separated is dissolved in a minimum amount of a suitable solvent and applied as a spot on the starting line with the help of a fine capillary or micro syringe.
- The spotted chromatographic paper is then suspended in a suitable solvent or a mixture of solvents. The position of the paper should be such that the spot on the starting line remains above the surface of the solvent. Thus, the solvent acts as the mobile phase.
- The solvent rises up the paper strip by capillary action and flows over the sample spot. The different components of the mixture depending upon their solubility (or partitioning between) in the stationary and mobile phases, travel through different distances.
- When the solvent reaches the finish line the paper strip is taken out and dried in air. The paper strip so developed is called a chromatogram.
- The spots of the separated coloured compounds are visible at different distances from their initial position on the starting line.
- The spots of the separated colourless compounds are observed either by placing the paper strip under UV light or by using an appropriate spray reagent as discussed in TLC.
- The components of the mixture are then identified by determining their R j values as discussed in TLC.
- The process can also be performed by folding the chromatographic paper into a cylinder

Criteria of purity of organic compounds
Several methods for the purification of organic compounds have already been discussed. The next important step is to test their purity, i.e., to know whether a particular compound has been purified or not. A pure organic solid has a definite and sharp melting point. An impure solid melts over a range of temperatures and even the presence of traces
Qualitative Analysis Of Organic Compounds
Since organic compounds are either hydrocarbons or their derivatives, the elements which occur in them are carbon (always present), hydrogen (nearly always present), oxygen (generally present), nitrogen, halogens, sulphur (less commonly present), phosphorus and metals (rarely present). All these elements can be detected by suitable methods.
1. Detection of carbon and hydrogen(C-H)
Detection of carbon and hydrogen Principle:
A small amount of a pure and dry organic compound is strongly heated with dry cupric oxide in a hard glass test tube when carbon present in it is oxidised to carbon dioxide and hydrogen is oxidised to water.
![]()
Liberated CO2 turns lime water milky and the liberated water vapours (H2O) which get condensed in the bulb of the delivery tube, turn white anhydrous CuSO4into blue hydrated copper sulphate (CuSO4-5H2O)
Ca(OH)2 (Lime Water)+ CO2→CaCO3↓ (Milky)+ H2O
CuSO4 Anhydrous (white)+ 5H2O→CuSO4.5H2O(Blue)
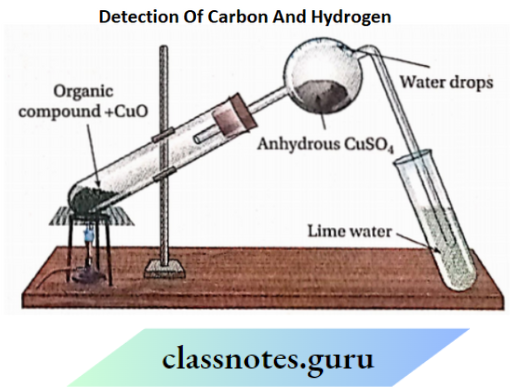
If the organic compound is a volatile liquid or gas, then the vapours of the compound are passed through heated copper oxide and the presence of C02 and H2O in the liberated gas can be proved in the same way.
If the organic compound contains sulphur in addition to carbon and hydrogen, then sulphur is oxidised to SO2 which also turns lime water milky due to the formation of calcium sulphite. In that case, the resulting gases are first passed through an acidified solution of potassium dichromate which absorbs SO2 and then through lime water.

2. Detection of nitrogen
1. Sodalime test
A very small amount of an organic compound is strongly heated with soda lime (NaOH + CaO) In a test tube. The evolution of ammonia having a typical smell Indicates the presence of nitrogen in the compound.
Example:
CH3CONH2 (Acetamide)+ [NaOH + CaO]→ CH3COONa (Sodium acetate)+ NH3↑
Sodalime test Limitation:
Organic compounds containing nitrogen as nitro ( — NO2) or azo (— N =N— ) groups do not evolve NH3 upon heating with soda lime, i.e., do not give this test.
2. Lassaigne’s test
Preparation of Lassaigne’s extract or sodium extract:
A pea-sized freshly cut dry sodium metal is heated gently in a fusion tube till it melts to a shining globule. A small amount ofthe organic compound is added and the tube is heated strongly for 2-3 minutes till it becomes red hot. The hot tube is then plunged into 10-15 mL of distilled water taken in a mortar. The mixture is then ground thoroughly by a pestle and filtered. The filtrate is known as sodium extract or Lassaigne’s extract.
Test for nitrogen:
The Lassaigne extract is usually alkaline in nature because the excess of metallic sodium reacts with water to form sodium hydroxide. A small amount of freshly prepared FeSO4 solution is added to a part of sodium extract and the contents are boiled when a light green precipitate of Fe(OH)2 is obtained.
The mixture is then cooled and acidified with dil.H2SO4. The immediate appearance of blue or green colouration or deep blue coloured precipitate of Prussian blue, Fe4[Fe(CN)6)3 indicates the presence of nitrogen.
Organic Compound Purification Methods Class 11 Notes
Chemistry of Lassaigne’s test:
1. When organic compound is fused with metallic Na, carbon and nitrogen present in it combine with Na to form NaCN
![]()
2. When the sodium extract is heated with FeSO4 solution, Fe(OH)2 is obtained which reacts with sodium cyanide to form sodium ferrocyanide. This is also obtained when FeSO4 reacts with NaCN.
FeSO4 + 2NaOH →Fe(OH)2 + Na2SO4
6NaCN + Fe(OH)2 →Na4 [Fe(CN)6 ] (Sodium ferrocyanide) + 2NaOH
FeSO4 + 6NaCN→Na2 SO4 + Na4 [Fe(CN)6 ]
Ferrous (Fe2+) ions present in hot alkaline solution undergo oxidation by O2 to give ferric (Fe3+) ions. These ferric ions then react with sodium ferrocyanide to produce ferric ferrocyanide (prussian blue)
4Fe(OH)2 + O2 + 2H2O→4 Fe(OH)3
2Fe(OH)3 + 3H2 SO4 →Fe2(SO4 )3 + 6H2O
3Na4 [Fe(CN)6] + 2Fe2(SO4)3→ Fe4[Fe(CN)6]3 (Prussian Blue)+ 6Na2SO4
1. In this test, it is desirable not to add FeCl3 solution because yellow FeCl3 causes Prussian blue to appear greenish. For the same reason, the alkaline extract should not be acidified with hydrochloric acid (which produces ferric chloride).
2. When the compound under investigation contains both nitrogen and sulphur, it may combine with sodium during fusion to form sodium thiocyanate (sulphocyanide). This gives blood red colouration with ferric chloride due to the formation of ferric thiocyanate. Thus, Prussian blue is not obtained.
Purification and Analysis of Organic Compounds Class 11 NCERT Notes
⇒ \(\mathrm{Fe}^{3+}+3 \mathrm{NaSCN} \rightarrow \mathrm{Fe}(\mathrm{SCN})_3+3 \mathrm{Na}^{+}\)
3. If fusion is carried out with excess of sodium, the resulting thiocyanate decomposes to give sodium cyanide and sodium sulphide. So, in that case no blood red colouration is visualised.
NaSCN + 2Na→NaCN + Na2S
However, in that case, black precipitate of FeS is obtained when FeS04 solution is added to sodium extract. The black precipitate dissolves on the addition of dil. H2SO4 and the test of nitrogen is then performed with that clear solution.
FeSO4 + Na2S→FeS↓ (Black) + Na2SO4
FeS + H2SO4 →H2S↑+ FeSQ4
Lassaigne’s test Limitations:
- Volatile organic compounds if present, escape before reacting when fused with metallic sodium.
- Some nitro compounds may lead to explosion during the fusion process.
3. Detection of sulphur
1. Lassaigne’s test:
Lassaigne’s extract is prepared as described in the case of nitrogen Sulphur present in the compound (which does not contain nitrogen) reacts with sodium metal to form sodium sulphide:

The following tests are then performed with the extract to detect the presence of sulphur
Lead acetate test:
One part ofthe extract is acidified with acetic acid and then a lead acetate solution is added to it. The formation of a black precipitate of lead sulphide (PbS) confirms the presence of sulphur in the compound
Na2S + Pb(CH3COO)2→PbS↓(Black) + 2CH3COONa
Sodium nitroprusside test:
A few drops of sodium nitroprusside solution are added to another part of the extract. The appearance of a violet or purple colouration confirms the presence of sulphur in the compound.
Na2S + Na2[Fe(CN)5NO](Sodium nitroprusside)→ Sodium sulphonitroprusside Na4[Fe(CN)5NOS](Violet)
2. Oxidation test:
The organic compound is fused with a mixture of potassium nitrate and sodium carbonate and as a result, sulphur, if present, gets oxidised to sodium sulphate.
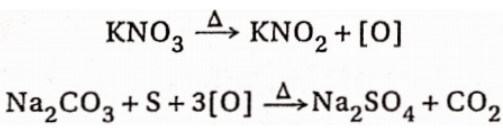
The fused mass is extracted with distilled water and filtered. The filtrate is acidified with dil. HCl and then barium chloride solution is added to it. The formation of a white precipitate insoluble in hydrochloric acid indicates the presence of sulphur in the compound
Na2SO4 + BaCl2→2NaCl + BaSO4↓ (White)
4. Detection of halogens
1. Beilstein’s test:
A clean and stout Cu-wire flattened at one end is heated in the oxidising flame of a Bunsen burner until it imparts any green or bluish-green colour to the flame. The hot end of the Cu-wire is then touched with the organic compound under investigation and is once again introduced into the flame. The reappearance of green or bluish-green flame due to the formation of volatile copperhalide indicates the presence of halogens in the compound.
Beilstein’s test Limitations:
Many halogen-free compounds,
For example:
Certain derivatives of pyridine and quinoline, purines, acid amides, urea, thiourea, cyano compounds etc. give this test presumably owing to the formation of volatile copper cyanides.
Therefore, this test is not always trustworthy. It does not indicate which halogen (Cl, Br or I) is present in the organic compound.
The test is not given by fluoro compounds since copper fluoride is non-volatile.
2. Lassaigne’s test:
Lassaigne’s extract is prepared as described in the case of nitrogen. During fusion, Na combines with the halogen present in the compound to form sodium halide

The extract is then boiled with dilute HNO3, cooled and a few drops of AgN03 solution are added to it White or yellow precipitation confirms the presence of halogen.
NaX + AgNO3 → AgX↓ + NaNO3 [X = Cl,Br,I]
1. Formation of a curdy white precipitate soluble in ammonium hydroxide solution indicates the presence of chlorine in the organic compound
NaCl + AgNO3→AgCU(White) + NaNO3
AgCl + 2NH4OH→[Ag(NH3)2]Cl(Soluble) + 2H2O
CBSE Class 11 Chemistry Organic Compound Analysis Summary
The formation of a pale yellow precipitate partially soluble in ammonium hydroxide solution indicates the presence of bromine in the organic compound.
NaBr + AgNO3→ AgBr4-(Pale yellow) + NaNO3
AgBr + 2NH4OH → [Ag(NH3)2]Br(Soluble) + 2H2O
The formation of a yellow precipitate insoluble in NH4OH solution indicates the presence of iodine in the organic compound.
Nal + AgNO3→AgI ↓ (Yellow) + NaNO3
Function Ol nitric acid:
If the organic compound contains nitrogen and sulphur along with halogens, Lassaigne’s extract contains sodium sulphide (Na2S) and sodium cyanide (NaCN) along with sodium halide (NaX). These will form a precipitate with silver nitrate solution and hence will interfere with the test.
Na2S + 2AgNO3→Ag2S↓ (Silver sulphide (Black) + 2NaNO3
NaCN + AgNO3→ AgCN↓ (Silver cyanide) (white)+ NaNO3
For this reason, before the addition of AgNO3 solution to the sodium extract for the detection of halogens, the extract is boiled with dilute HNO3 which decomposes sodium sulphide and sodium cyanide to vapours of H2S and HCN respectively
Alternatively, sulphide and cyanide ions can be removed by adding 5% nickel (II) nitrate solution which reÿct? with these ions forming precipitates of nickel (II) sulphide and nickel (II) cyanide
3. Chlorine water test for bromine and iodine:
- A portion of Lassaigne’s extract is boiled with dilute nitric acid or dilute sulphuric acid to decompose NaCN and Na2S.
- The solution is then cooled, and acidified with dil. H2SO4 and a few drops of carbon disulphide or carbon tetrachloride solution are added to it.
- The resulting mixture is then shaken with a few drops of freshly prepared chlorine water and allowed to stand undisturbed for some time.
An orange or brown colouration in the carbon disulphide or carbon tetrachloride layer confirms the presence of bromine, whereas a violet colouration in the layer confirms the presence of iodine in the compound
2NaBr + Cl2→2NaCl + Br2 (turns CS2 or CCl4 layer orange)
2NaI + Cl2→2NaCl + I2 (turns CS2 or CCl4 layer violet)
5. Detection of phosphorus
1. The organic compound under investigation is fused with sodium peroxide (an oxidising agent) when phosphorus is oxidised to sodium phosphate

2. The fused mass is then extracted with water, filtered and the filtrate is boiled with concentrated nitric acid. The mixture is then cooled and an excess of ammonium molybdate solution is added to it
3. The appearance of a yellow precipitate or colouration due to the formation of ammonium phosphomolybdate, (NH4 )3 PO4 -12MoO3, confirms the presence of phosphorus in the compound.

Class 11 Chemistry Purification Techniques for Organic Compounds
Quantitative Analysis Of Organic Compounds
After detecting the presence of various elements in a particular organic compound, the next step is to determine their respective percentages. This is known as quantitative analysis.
1. Estimation of carbon and hydrogen
Both carbon and hydrogen present in an organic compound are estimated by Liebig’s method.
Liebig’s method
Liebig’s method Principle:
A pure and dry organic compound of known mass is heated strongly with pure and dry copper oxide (CuO) in an atmosphere of air or oxygen-free from carbon dioxide. Both carbon and hydrogen present in the organic compound undergo complete oxidation and get converted into carbon dioxide and water respectively

COthus produced is absorbed in a previously weighed potash bulb containing a strong KOH solution while water produced is absorbed in a previously weighed U-tube containing anhydrous CaCl2. The U-tube and the bulb are weighed again and from the difference between the two weights, the amount of CO2 and HaO are determined.
Liebig’s method Procedure:
- The apparatus for the estimation of C and H is The apparatus consists of the following units:
- Combustion tube,
- U-tube containing anhydrous CaCl2 and
- a bulb containing strong KOH solution.
- The tube is heated l strongly for 2-3 hours till the whole of the organic compound is burnt up.
- After combustion is over, the absorption units (the Utube and the potash bulb) are disconnected and weighed separately

Results and calculations:
Let the mass of organic compound taken = w g. The increase in the mass of the potash bulb, i.e., the mass of CO2 formed = x g.
The increase in the mass of the U-tube, i.e., the mass of water formed =y g.
Percentage ot carbon: 1 mol of CO2 (44g) contains 1 gram atom of carbon (12 g).
∴ x g of CO2 contains = \(\frac{12x}{44}\) g of C
Now, \(\frac{12x}{44}\) g carbon is present in w g organic compound.
∴ The percentage of carbon in the compound
= \(\frac{2 y \times 100}{18 w}=\frac{2}{18} \times \frac{\text { Mass of } \mathrm{H}_2 \mathrm{O} \text { formed }}{\text { Mass of the compound taken }} \times 100\)
Percentage ot hydrogen: 1 mol of water (18g) contains 2 gram-atom hydrogen (2 g).
∴ y g of H2 O contains = \(\frac{2y}{18}\) g of H
Now, \(\frac{2y}{18}\) hydrogen is present in w g organic compound.
∴ The percentage of hydrogen in the compound
Estimation of C and H by Liebig’s method is suitable for organic compounds containing C, H and O only, but it requires some modifications for compounds containing nitrogen, halogens and sulphur.
1. Compounds containing nitrogen:
During combustion, N present in the organic compound undergoes oxidation to give its oxides (NO, NOz etc.) which are
Also absorbed in an alkali solution along with CO2. Oxides of nitrogen are decomposed back to nitrogen by placing a copper gauze roll near the exit of the tube. The N2 so produced is not absorbed by the alkali solution.
2. Compounds containing halogens:
During combustion, halogens present in the organic compound get converted into volatile copper halides which partly decompose to give free halogens. These halogens and volatile copper halides get dissolved in an alkali solution. This can be prevented by placing a silver gauze roll near the exit of the combustion tube. Halogens combine with silver to give non-volatile silver halides.
2Ag + X2 →2AgX; 2Ag + CuX2 → 2AgX + Cu
3. Compounds containing only sulphur or sulphur and halogen:
During combustion, elemental sulphur present in the organic compound is oxidised to SO2 which is absorbed in the potash bulb.
This can be prevented by placing a layer of fused lead chromate near the exit of the tube. SO2 combines with lead chromate to produce non-volatile lead sulphate.

Lead chromate also reacts with copper halides and free halogens to form lead halides which remain in the combustion tube

Liebig’s method Precautions:
- All the joints ofthe combustion must be air-tight.
- The combustion must be free from CO2 and water vapour.
- The airflow is controlled in such a way that only 2-3 bubbles are generated per second. A faster flow of air may lead to the formation of carbon monoxide.
- If carbon is deposited on the surface of the combustion mbe, then oxygen instead of air is to be passed at the final stage ofthe process
Class 11 Chemistry Purification Techniques for Organic Compounds
Example 1: 0.90g of an organic compound on complete combustion yields 2.20 g of carbon dioxide and 0.60 g of water. Calculate the percentages of carbon and hydrogen in the compound.
Answer:
Mass of the organic compound = 0.90 g
Mass of CO2 formed = 2.2 g & mass of H2O formed = 0.6 g
Percentage of carbon: 44 g CO2 contains = 12 g carbon
0.90 g organic compound contains = \(\frac{12 \times 2.20}{44}\) g of carbon
0.20 g organic compound contains = \(\frac{12 \times 2.20}{44}\) g carbon
Percentage of carbon = \(\frac{12 \times 2.20 \times 100}{44 \times 0.90}\)
= 66.67
Percentage of hydrogen: 18 g of H2O contains = 2g of hydrogen
0.60 g of H2O contains = \(\frac{2 \times 0.60}{18}\) of hydrogen
0.90 g compound contains = \(\frac{2 \times 0.60}{18}\) g hydrogen
Percentage of hydrogen = \(\frac{2 \times 0.60 \times 100}{18 \times 0.90}\)
= 7.41
2. Estimation ot nitrogen
The following two methods are commonly used for the estimation of nitrogen in an organic compound
- Duma’s method
- Kjeldahl’s method.
1. Dumas method
Dumas method Principle:
A weighed amount of the organic compound is heated with an excess of copper oxide in an atmosphere of CO2. Carbon and hydrogen present in the compound are oxidised to CO2 and H2O respectively while nitrogen is set free as nitrogen gas (N2). If any oxide of nitrogen is formed during this process, it is reduced back to nitrogen by passing over hot reduced copper gauze

When the gaseous mixture thus obtained is passed through a 40% KOH solution taken in a Schiff’s nitrometer, all gases except nitrogen are absorbed by KOH. The volume of nitrogen collected over the KOH solution is noted and from this, the percentage of nitrogen in the compound is calculated

Dumas method Procedure:
- The apparatus used for the estimation of nitrogen by the Dumas method ). It consists of:
- CO2 generator combustion tube (a glass tube of diameter 15m/r and length 90 cm) and Schiff’s nitrometer.
- The combustion tube is packed with [a] coarse CuO which prevents backward diffusion of gases produced,
- an accurately weighed amount of organic compound of approximately 0.2g mixed with excess of CuO.
- Coarse CuO and
- A reduced copper gauze can reduce any oxides of nitrogen back to N2.
- The Schiff’s nitrometer (connected to the combustion tube) consists of a graduated tube with a reservoir and a tap at the upper end. It has a mercury seal at the bottom to check the backward flow ofthe KOH solution.
- CO2 generated by heating NaHCO3 or MgCO3 is dried by passing through concentrated HS04 and then passed through the combustion tube to displace the air present in the tube.
- The combustion tube is then heated in the furnace. When the combustion is complete, a rapid stream of CO2 is passed through the tube to sweep away the last traces of N2.
- The volume of nitrogen collected over the KOH solution in the nitrometer tube is recorded after careful levelling (by making the level of KOH solution both in the tube and reservoir the same).
- The room temperature is recorded and the vapour pressure of the KOH solution at the experimental temperature is noted in the table.
Results and calculations:
Let the mass of the organic compound taken = w g, the volume of N2 gas collected = V mL, the atmospheric pressure = P mm, the room temperature = t°C and the vapour pressure of KOH solution at t°C =f mm. Hence, the pressure of dry N2 gas = (P- f) mm.
By applying the gas equation = \(\frac{P_1 V_1}{T_1}=\frac{P_2 V_2}{T_2}\)
Volume of N2 at STP, = \(V_2=\frac{P_1 V_1 T_2}{P_2 T_1}=\frac{(P-f) \times V \times 273}{760(273+t)}\)
Percentage of nitrogen: 22400 mL of N2 gas at STP will weigh = 28 g.
2 mL of N2 gas at STP will weigh = \(\frac{28 \times V_2}{22400} \mathrm{~g}\)
Percentage of nitrogen:
= \(\frac{\text { Mass of nitrogen }}{\text { Mass of the compound }} \times 100\)
= \(\frac{28}{22400} \times \frac{V_2}{w} \times 100\)
The amount of nitrogen estimated by this method is 0.2% higher than the actual quantity. This is because a fraction of the small amount of air (nitrogen) entrapped in copper oxide gets deposited in the nitrometer
Example:
In Dumasg of an method organic for compound, the estimation gave 31.7 of nitrogen, mL of nitrogen at 14°C and 758 mm pressure. Calculate the percentage of nitrogen in the compound (vapour pressure of water at 14°C = 12 mm.
Answer:
If the volume ofnitrogen at STP is V mL, then
V = \(\frac{31.7(758-12)}{(273+14)} \times \frac{273}{760}\)
= 29. 6 mL
Now, at STP 22400 mL of nitrogen will weigh = 28 g
∴ 29.6 mL nitrogen at STP will weigh = \(\) = 0.037%
∴ Percentage of nitrogen = \(=\frac{0.037 \times 100}{0.1877}\)
= 19.71
2. Kjeldahl’s method
Kjeldahl’s method Principle:
A known mass of a nitrogenous organic compound is digested (heated strongly) with concentrated H2SO4 in the presence of a little potassium sulphate (to increase the boiling point of H2SO4) and a little CuSO4 (a catalyst) when the nitrogen present in the compound is quantitatively converted into ammonium sulphate. The resulting mixture is heated with excess of NaOH solution and the ammonia evolved is passed into a known but excess volume of a standard acid (HCl or H2SO4 ).

The excess acid left after the neutralisation of ammonia is estimated by titration with some standard alkali
2NaOH + H2SO4 →Na2SO4 + 2H2O
Kjeldahl’s Method Procedure
- The organic compound (0.5-5 g) is heated with a cone. H2SO4 in a Kjeldahl’s flask which is a long-necked round-bottomed flask with a loose stopper. A small amount of potassium sulphate and a few drops of mercury (or a little CuS ) are added.
- The reaction mixture is heated for 2-3 hours when carbon and hydrogen present in the compound are oxidised to CO2 and H2O respectively while nitrogen is quantitatively converted to ammonium sulphate.
- CO2 and water vapours escape through the loose stopper while ammonium sulphate remains in the flask.
- The Kjeldahl’s flask is then cooled and the contents of the flask are transferred to a round-bottomed flask.
- The mixture is diluted with water and excess caustic soda solution (40%) is added.
- The flask is then connected to a Liebig condenser through Kjeldahl’s tap. The lower end of the condenser is dipped in a known volume of standard acid (N/10 HCl or H2SO4 ) taken in a conical flask.
- The flask is then heated. The evolved ammonia gas is absorbed in the acid solution.
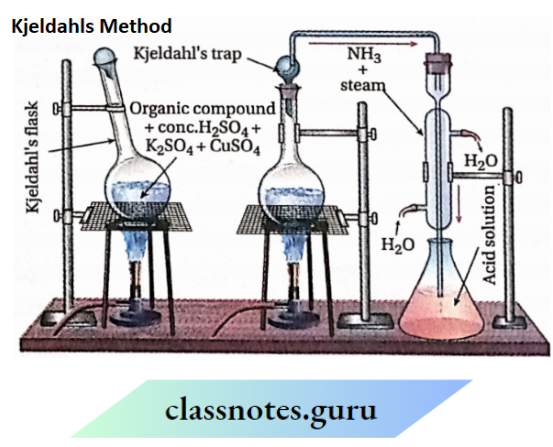
Results and calculations:
Let the mass of the organic compound = w g, volume of acid taken = V1 mL, normality of acid solution x(N) and volume of x(N) alkali required to neutralise unused acid = V2 mL.
Now, V2 mL × (N) alkali = V2 mL × (N) acid.
∴ The quantity of acid used for neutralising ammonia = VjmL x(N) acid solution- V2 mL x(N) acid solution = (V1– V2) mL x(N) acid solution = (V1– V2)mL x(N) ammonia solution.
Now, 1000 mL (N) ammonia solution contains 17 g of NH3 or 14 g of nitrogen.
Therefore, (V1– V2)mL × (N)
Ammonia solution contains = \(\frac{14 \times\left(V_1-V_2\right) x}{1000}\) g of nitrogen
This amount of nitrogen is present in w g compound
% of nitrogen = \(\mathrm{n}=\frac{14 \times\left(V_1-V_2\right) x \times 100}{1000 \times w}=\frac{1.4\left(V_1-V_2\right) x}{w}\)
= \(\frac{1.4 \times \text { Vol. of acid used } \times \text { Normality of the acid used }}{\text { Mass of the compound taken }}\)
Kjeldahl’s method Limitations:
- Nitrogen present in pyridine, quinoline, diazo compound, azo compound, and nitro compound, cannot be converted into ammonium salts by this method.
- So this method does not apply to such compounds. Thus although Dumas’s method applies to all compounds, Kjeldahl’s method has limited applications.
The percentage of nitrogen estimated in this method is not correct.
Kjeldahl’s method Utility:
This experiment can be performed quite easily and quickly. So in those cases where correct estimation of nitrogen content is not necessary
For example: Fertiliser, soil, food¬ stuff etc.), this method has its application.
This method is not troublesome and hence the possibility of error can be minimised by repeating the experiment several times.
NCERT Solutions Class 11 Chemistry Purification and Analysis of Organic Compounds
Example: 0.257 g of a nitrogenous organic compound was analysed by Kjeldahl’s method and ammonia evolved was absorbed in 50 mL of (N/10) H2SO4. The unused acid required 23.2 mL of (N/10) NaOH solution for complete neutralisation. Determine the percentage of nitrogen in the compound.
Answer:
Acid taken = 50 mL (N/10) H2SO4 solution
= 5 mL(N) H2SO4 solution
Alkali required to neutralise the excess acid
= 23.2 mL (N/10) NaOH = 2.32 mL (N) NaOH
Now, 2.32 mL(N) NaOH = 2.32 mL(N) H2SO4 solution
The acid required to neutralise the ammonia evolved
= 5 mL (N) H2SO4– 2.32 mL (N) H2SO4 solution
= 2.68 mL (N) H2SO4 s 2.68 mL (N) NH3
[v KmL(N) H2SO4S VmL(N) ammonia]
1000 mL (N) ammonia solution contains 14g nitrogen
So, 2.68mL (N) ammonia solution contains \(\frac{14 \times 2.68}{1000}\) g nitrogen
So, 0.257g compound contains = \(\frac{14 \times 2.68}{1000}\) g nitrogen
Percentage of nitrogen = \(=\frac{14 \times 2.68 \times 100}{1000 \times 0.257}\)
= 14.6
3. Estimation of halogens: Carius method
Halogens Carius method Principle:
An organic compound containing halogen of known mass is heated with fuming nitric acid and a few crystals of silver nitrate. The halogen present in the compound is converted into the corresponding silver halide (AgX). From the mass of the compound taken and that of the silver halide formed, the percentage of halogen in the compound can be calculated.
Carius method Procedure:
- About 5 mL of fuming nitric acid and 2 g of silver nitrate crystals are placed in a hard glass tube (Carius tube) of about 50 cm in length and 2 cm in diameter.
- A small amount of accurately weighed organic compound is taken in a small tube and the tube is placed carefully into the Carius tube in such a way that nitric acid does not enter the tube.
- The Carius tube is then sealed. Now, it is placed in an outer iron jacket and heated in a furnace at 550-560 K for about six hours
- As a result, C, H and S present in the compound are oxidised to CO2, H2O and H2SO4 respectively. The halogen present in the compound gets converted into silver halide which is precipitated.
- The tube is cooled and when the sealed capillary end is heated in a burner’s flame, a small hole is formed through which the gases escape.
- The capillary end is now cut off and the precipitate of silver halide is filtered, washed, dried and weighed
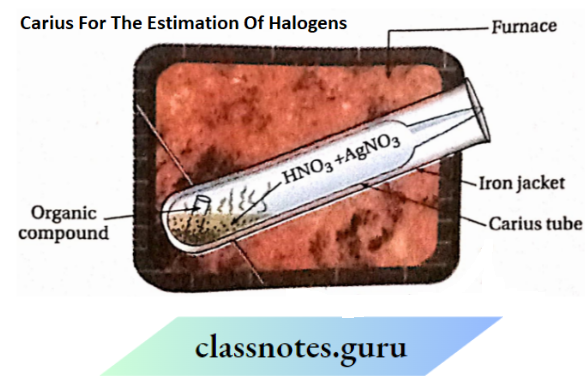
Results & calculations:
Let, the mass of organic compound taken = w g and the mass of silver halide (AgX) formed—x g. Now, 1 mol of AgX contains 1 gram-atom of X (X = Cl, Br or I ), i.e., (108 + atomic mass of X) g of AgX contain (atomic mass of X)g of X
∴ xg AgX contain = \(\frac{\text { atomic mass of } X}{(108+\text { atomic mass of } X)}\) X JCg of X
This amount of halogen (X) is present in w g compound.
Percentage of halogen in the compound
= \(\frac{\text { atomic mass of } \mathrm{X}}{(108+\text { atomic mass of } \mathrm{X})} \times \frac{x}{w} \times 100\)
1. Percentage of chlorine (atomic mass = 35.5 )
= \(\frac{35.5}{(108+35.5)} \times \frac{x}{w} \times 100\)
= \(\frac{35.5}{143.5} \times \frac{x}{w} \times 100\)
2. Percentage of bromine (atomic mass = 80 )
= \(\frac{80}{(108+80)} \times \frac{x}{w} \times 100\)
= \(\frac{80}{188} \times \frac{x}{w} \times 100\)
3. Percentage of iodine (atomic mass = 127 )
= \(\frac{127}{(108+127)} \times \frac{x}{w} \times 100\)
= \(\frac{127}{235} \times \frac{x}{w} \times 100\)
NCERT Solutions Class 11 Chemistry Purification and Analysis of Organic Compounds
Example: In the Carius method, 0.256 g of an organic compound containing bromine gave 0.306 g of AgBr. Find out the percentage of bromine in the compound.
Answer:
The mass of the organic compound taken = 0.256 g and the mass of AgBr formed = 0.306 g.
Now, 1 mol of AgBr = 1 gram-atom of Br
or, (108 + 80) g or 188 g of AgBr = 80 gofBr
i.e., 188 g of AgBr contains = 80 g of bromine
0.306 g of Ag Br contain \(\frac{80}{188} \times 0.306\) g of bromine
This amount of bromine is present in 0.256g compound
Percentage6 of bromine = \(\frac{80}{188} \times \frac{0.306}{0.256} \times 100\)
= 50.9
4. Estimation of sulphur: Carius method
Sulphur Carius method Principle:
When an organic compound containing sulphur is heated with fuming nitric acid in a sealed tube (Carius tube), sulphur is quantitatively oxidised to sulphuric acid. It is then precipitated as barium sulphate by adding a barium chloride solution. The precipitate is then filtered, washed, dried and weighed. From the amount of BaSO4 formed, the percentage of sulphur is calculated.

Resultsandcalculations: Let the mass of organic compound
= w g and the mass of barium sulphate formed = xg
Now, 1 mol of BaSO4 contains 1 gram-atom of S, i.e.,
(137 + 32 + 64) g or, 233 g BaO4 contain 32 g sulphur.
∴ x g of BaSO4 contain \(\frac{32}{233}\) × x g of sulphur
So, this amount of sulphur is present in the w g compound.
∴ Percentage of sulphur = \(\frac{32}{233} \times \frac{x}{w} \times 100\)
Example: In sulphur estimation by the Carius method, 0.79 g of an organic compound gave 1.164 g of barium sulphate. Calculate the percentage of sulphur in the compound.
Answer:
The mass of the organic compound taken = 0.79 g and the mass of BaSO4 obtained = 1.164 g.
Now, 1 mol of BaSO4 = 1 gram-atom of S or,
233 g of BaSO4= 32 g of i.e.,
233 g of BaSO4 contain = 32 g ofsulphur.
1.164 g of BaSO4 contain \(\frac{32}{233} \times 1.164\) × 1.164 g of sulphur
So, 0.79 g compound contain \(\frac{32}{233} \times 1.164\) × 1.164 g of sulphur
Percentage of sulphur = \(\frac{32}{233} \times \frac{1.164}{0.79} \times 100\)
= 20.24
5. Estimation of phosphorus: Carius method
Phosphorus Carius method principle:
An organic compound (containing P) of known mass is heated with fuming nitric acid in a sealed tube (Carius tube). Phosphorus present in the compound is oxidised to phosphoric acid which is precipitated as ammonium phosphomolybdate by heating it with concentrated nitric acid and then adding ammonium molybdate.

Ammonium phosphomolybdate (Yellow)
The precipitate of ammonium phosphomolybdate thus obtained is filtered, washed, dried and weighed.
Alternative method:
The phosphoric acid is precipitated as magnesium ammonium phosphate (MgNH4PO4) by the addition of magnesia mixture (a solution containing MgCl2, NH4Cl and NH4OH ).

The precipitate is filtered, washed, dried and then ignited to give magnesium pyrophosphate (Mg2P2O)

From the mass of magnesium pyrophosphate, the percentage of phosphorus in the compound can be easily calculated.
Results and calculations:
Let the mass of the organic compound taken = wg and the mass of ammonium phosphomolybdate formed = xg.
1 mol (NH4)3PO4-12MoO3 contains 1 gram-atom of P.
or, 3(14 + 4) + 31 + 4 × 16 + 12(96 + 3 × 16) = 1877g of
(NH4)3PO4. 12MOO3 = 31 g of P, i.e., 1877 g of
(NH4)3PO4 . 12MoO3 contain 31 g of phosphorus
xg of (NH4)3PO4 -12MoO3 contain \(\)
xg of phosphorus. This amount of phosphorus is present in w g of the compound.
Percentage of phosphorus = \(\frac{31}{1877} \times \frac{x}{w} \times 100\)
= \(\frac{31}{1877} \times \frac{\text { Mass of ammonium phosphomolybdate }}{\text { Mass of the compound taken }} \times 100\)
Alternative calculation: Let the mass of Mg2P20? formed
= xg. Now, 1 mol of (Mg2P2O) contains 2 gram-atom of P
or, (24 × 2 + 31 × 2 + 16 × 7) = 222 g of (Mg2P2O)
i.e., 222g of (Mg2P2O) contains 62 g of phosphorus.
x g of (Mg2P2O)contain \(\frac{62}{222} \times x\) g of phosphorus
w g organic compound contain \(\frac{62}{222} \times x\) xg phosphorus
Percentage of phosphorus = \(\frac{62}{222} \times \frac{x}{w} \times 100\)
= \(\frac{62}{222} \times \frac{\text { Mass of } \mathrm{Mg}_2 \mathrm{P}_2 \mathrm{O}_7 \text { formed }}{\text { Mass of the compound taken }} \times 100\)
NCERT Solutions Class 11 Chemistry Purification and Analysis of Organic Compounds
Example: 0.35 g of an organic compound containing phosphorus gave 0.65 g magnesium pyrophosphate, Mg2P2O in the Carius method. Calculate the percentage of phosphorus in the given compound.
Answer:
The mass of the organic compound taken = 0.35 g and the mass of Mg2P2O formed = 0.65 g.
Now, 1 mol of Mg2P2O contains 2 gram-atom of P.
Or, (2 × 24 + 2 × 31 + 16 × 7) g of Mg2P2O= 2 × 31 g of P
i.e., 222 g of Mg2P2O contains 62 g of phosphorus
0.65 g Mg2P2Ocontain \(\frac{62}{222} \times 0.65\) x 0.65 g of phosphorus
This amount of P is present in a 0.35 g compound.
Percentage of phosphorus = \(\frac{62}{222} \times \frac{0.65}{0.35} \times 100\)
6. Estimation of oxygen
Percentage of oxygen = 100 – (sum of the percentages of all other elements). However, the oxygen content of a compound can be estimated directly as follows:
Oxygen Principle:
An organic compound of known mass is decomposed by heating in a stream of nitrogen gas. The mixture of the gaseous products including oxygen is passed over red-hot coke when all the oxygen combines with carbon to form carbon monoxide. The mixture is then passed through warm iodine pentoxide (I2O5 ) when CO undergoes oxidation to CO2 and iodine is liberated.

From the knowledge of the mass of iodine liberated or C02 produced, the percentage of oxygen in the compound can be calculated easily
Results and calculations: Let, the mass of the organic compound taken = wg and the mass of CO2 formed =xg. We find that each mole of oxygen liberated from the compound will produce 2 mol of CO2
x g. of CO2 is obtained from \(\frac{32}{88} \times x=\frac{16}{44} \times\) = x x g of O2
This amount of oxygen is present in w g ofthe compound.
Percentage of oxygen = \(\frac{16}{44} \times \frac{x}{w} \times 100\)
1. Percentage of carbon (Liebig’s method)
= \(\frac{12}{44} \times \frac{\text { Mass of } \mathrm{CO}_2 \text { formed }}{\text { Mass of the compound taken }} \times 100\)
2. Percentage of carbon (Liebig’s method)
= \(\frac{2}{18} \times \frac{\text { Mass of } \mathrm{H}_2 \mathrm{O} \text { formed }}{\text { Mass of the compound taken }} \times 100\)
3. Percentage of nitrogen (Dumas method)
= \(\frac{28}{22400} \times \frac{\text { Vol. of } \mathrm{N}_2 \text { gas at STP }}{\text { Mass of the compound taken }} \times 100\)
4. Percentage of nitrogen (KjeldahTs method)
= \(\frac{28}{22400} \times \frac{\text { Vol. of } \mathrm{N}_2 \text { gas at STP }}{\text { Mass of the compound taken }} \times 100\)
5. Percentage of halogen (Carius method)
= \(\frac{\text { At. mass of } X}{(108+\text { At. mass of } X)} \times \frac{\text { Mass of AgX }}{\text { Mass of the compound taken }} \times 100\)
X = Cl(35.5) , Br(80) or 1(127)
6. Percentage of sulphur (Carius method)
= \(\frac{32}{233} \times \frac{\text { Mass of } \mathrm{BaSO}_4 \text { formed }}{\text { Mass of the compound taken }} \times 100\)
7. Percentage of phosphorus (Carius method)
\(\frac{62}{222} \times \frac{\text { Mass of } \mathrm{Mg}_2 \mathrm{P}_2 \mathrm{O}_7 \text { formed }}{\text { Mass of the compound taken }} \times 100\)8. Percentage of oxygen
= \(\frac{16}{44} \times \frac{\text { Mass of } \mathrm{CO}_2 \text { formed }}{\text { Mass of compound taken }} \times 100\).