
Fundamental Concepts Of Organic Reaction Mechanism
In an organic reaction, the organic molecule (called the substrate) reacts with a suitable attacking species (called the reagent) to form products. The formation of produces) may take place either directly from the reactants (i.e., substrate and reagent) through a transition state or the formation of one or more intermediates. Some by-products may also be formed from the intermediate

The reagents are mostly either positively or negatively charged. A positively charged reagent attacks the site which is rich in electrons while a negatively charged reagent attacks that site which is electron deficient. So, for the reaction to take place in the covalent bond of the substrate, the bond must have some degree of ionic character.
Although the bond of an organic compound is mainly covalent, that bond becomes partially ionic due to some permanent or temporary displacement of the bonding electrons.
The ionic nature of a bond may be attributed to the following reasons:
- Inductive effect
- Electromeric effect
- Resonance
- Hyperconjugation.
Besides these electronic effects, steric effect or steric hindrance plays a very important role in determining the reactivity of organic compounds
Read and Learn More CBSE Class 11 Chemistry Notes
1. Inductive effect
Inductive effect Definition:
The permanent displacement of electrons along a carbon chain which occurs when some atom or group, either more or less electronegative than carbon is attached to the carbon chain is called the inductive effect
Inductive effect When a covalent bond is formed between two atoms having different electronegativities, the bonding electron pair is not shared equally by the two atoms. The electron pair being attracted by the more electronegative atom gets shifted more towards it. Consequently, the more electronegative atom acquires a partial negative charge (i.e., 6- ) and the less electronegative atom acquires a partial positive charge (Le., d+).
Organic Reaction Mechanism Fundamentals Class 11 Notes
Example:
When an electronegative (electron-withdrawing) Clatom (or a group such as — NO2 ) is attached to the end of a carbon chain (whose carbon atoms are designated as 1, 2, 3,… etc.), the cr -electrons ofthe Cx —Cl bond are attracted by or displaced more towards the Cl-atom. As a result, the Cl-atom acquires a partial negative charge (δ-) and the carbon, C1, acquires a partial positive charge (δ+). As Cj is now somewhat positively charged, it in turn, attracts the cr -electrons of the C1-C2 bond towards it So, C2 acquires a partial positive charge (SS+) smaller in magnitude than that on C1. Similarly, C3 acquire a partial positive charge (δδδ+) even smaller than that on C2

Similarly, if an element less electronegative than carbon, such as lithium (Li) (or any other electron-releasing group or atom), is attached to the terminal C -atom bf a carbon chain, then a partial positive charge (<5+) is developed on the Li-atom and a partial negative charge (6-) is developed on the Cx -atom.
The small negative charge on C1, in turn, repels the cr -electrons of the C1—C2 bond towards C2. As a result, C2 acquires a partial negative charge (δδ-) smaller in magnitude than that of C1 Similarly, C3 acquires a partial negative charge (δδδ-) which is even smaller than that of C2

Inductive effect Characteristics:
- This type of charge dispersal, which diminishes rapidly as the distance from the source increases, almost becomes negligible after the third carbon atom and is ignored.
- It is to be noted that although the inductive effect causes some degree of polarity in covalent bonds, the bond is never cleaved due to the effect.
- The inductive effect is represented by the symbol ( —<—). The arrow always points towards the more electronegative atom or group.
Measurement of inductive effect:
The inductive effect is always transmitted along a chain of carbon atoms. It cannot be expressed by any absolute value. The relative inductive effect of an atom or group is measured by taking H -atom of the R3C — H molecule as standard. When an atom or group- Z of the C — Z bond of the R3C —Z molecule attracts the bonding electrons more strongly than hydrogen of the C—H bond in the R3C— H molecule.
Then according to the definition introduced by Ingold, Z is said to have a negative inductive effect or electron-withdrawing inductive effect or -I effect. On the other hand, if the atom or group Z attracts the bonding electrons of the C — Z bond less strongly than the hydrogen atom of the C — H bond, then it is said to have a positive inductive effect or electron-releasing inductive effect or +1 effect
+I effect: —NH– > —O– > — COO– > (CH3)3C— > (CH3)2CH — > CH3CH2 — > CH3 — > D
– I effect: — +NR3> — +SR2> — +NH3 > — NO2 > -SO2R > — CN > — COOH > — F > — Cl > — Br > — I > — OR > — OH
Impact of inductive effect and its explanation:
Some important properties of organic compounds such as acidic property, basic property, bond polarity, and chemical reactivity vary remarkably due to inductive effect. Some examples of the influence of + 1 and – I effects on the properties of organic compounds are discussed here
1. Strength of monocarboxylic acids
Rule 1:
The relative strengths of monocarboxylic acids can be explained by the inductive effect ofthe substituent present in the carbon chain. If an electron releasing group or atom is attached directly to the -COOH group or to the carbon chain close to the -COOH group, then the positive inductive effect (+1) of such group increases the electron density on the oxygen atom of the
O— H group and consequently, the shared pair of electrons of the O — H bond is less strongly attracted towards the oxygen atom. As a result, the dissociation of the O —H bond to give H+ ion is less favoured. Thus, a group having a + I effect, when present in a monocarboxylic acid molecule decreases the strength of that acid.
On the other hand, if an electron-withdrawing group or atom is attached to the carbon chain close to the —COOH group, the negative inductive effect (-1) of such group decreases the electron density on the oxygen atom of the O— H group and consequently, the shared pair of electrons of O —H bond are more strongly attracted towards the oxygen atom. As a result, the dissociation of the O—H bond to give H+ ion is facilitated. Thus, a group having – I effect, when present in a monocarboxylic acid molecule, increases the strength of that acid molecule.
Example: Chloroacetic acid is stronger than acetic acid

Rule 2:
The strength of carboxylic acid increases as the extent of the effect ofthe substituent increases.
Example:
-I effect of the halogens follows the order: fluorine > chlorine > bromine > iodine. So, among the halogen-substituted acetic acids, trifluoroacetic acid (FCH2COOH) is the strongest while iodoacetic acid (ICH2COOH) is the weakest

Rule 3:
With the increase in the number of electron-attracting substituents, the strength of the acid increases.
Example:
Dichloroacetic acid is stronger than monochloroacetic acid while trichloroacetic acid is stronger than dichloroacetic acid.

Rule 4:
As the distance ofthe electron attracting substituent from the carboxyl group increases, the strength of the acid decreases.
Example:
2-chlorobutanoic acid is a stronger acid than 3- chlorobutanoic acid which in turn is stronger than 4- chlorobutanoic acid

Acid strength can also be explained in terms of the relative stabilities of the acid and its conjugate base. Electron withdrawing groups (EWG) disperse the negative charge on the anion [i.e., conjugate base), thus stabilising it and hence increasing acidity. On the contrary, electron-donating groups EDG intensify the negative charge on the anion, thus destabilise it and hence decrease acidity

2. The Basic Strength Of Amines
The increase in the strength of nitrogenous bases, e.g., amines, is related to the readiness with which they are prepared to take up protons and therefore, to the availability of the unshared pair ofelectrons on nitrogen.
Class 11 Chemistry Organic Reaction Mechanism NCERT Notes
Example:
We might expect the order of basic strength:
NH3<CH3NH2<(CH3)2NH<(CH3)3N> due to the increasing inductive effect (+1) of successive —CH3 groups making the N -atom more negatively charged, i.e., making the unshared pair of electrons more readily available. However, this sequence of basic strength of amines agrees with the results if measurements of basicity are made in the gas phase or in a solvent in which H -H-bonding does not take place

Basic strength increases (in the gas phase or in a solvent which does not form H-bond with amines). The introduction of electron-withdrawing groups close to the basic centre causes a decrease in F3Cthe basicity, due to their electron- Tri-trifluoromethylamine (virtually non-basic) withdrawing inductive effect.
An interesting example is the amine, (CF3)3N which is found to be virtually non-basic, due to the presence of three powerful electron-withdrawing — CF3 groups, each of which contains three highly electronegative F -atoms.

The order of basic strength of amines in aqueous medium is:
⇒ \(\left(\mathrm{CH}_3\right)_2 \ddot{\mathrm{N}} \mathrm{H}\left(2^{\circ}\right)>\mathrm{CH}_3 \ddot{\mathrm{N}} \mathrm{H}_2\left(1^{\circ}\right)>\left(\mathrm{CH}_3\right)_3 \ddot{\mathrm{N}}\left(3^{\circ}\right)\)
Due to the combined effect of hydrogen bonding and +1 effect of— CH3, groups, the conjugate acid of (CH3)2 NH i.e., (CH3)2 +NH2 is the most stable while the conjugate acid of (CH3)3N, i.e., (CH3)3 NH is the least stable and for this reason, in the aqueous medium, the above order of basicity observed
2. Electromeric effect
The complete transfer of a pair of 7t -electrons of a multiple bond (double bonds such as C=C, C=0 and triple bonds such as C = C and G= N ) to one of the multiple bonded atoms (usually the more electronegative one) in the presence of an attacking reagent is called electromeric effect or E-effect.
The transfer of the electron pair is indicated by a curved arrow. As soon as the reagent is removed, this effect vanishes and the molecule reverts back to its original position. Since this effect occurs by the presence ofthe attacking reagent, it takes place in the direction which facilitates the reaction. The electromeric effect may be represented as follows

Types of electromeric effect:
+ E-effect:
If the electron pair of the -bond is transferred to that doubly bonded atom to which the attacking species gets finally attached, then the effect is called +E-effect

– E-effect:
If the electron pair of the π-bond is transferred to that doubly bonded atom to which the attacking species Mous Lewis structures, which differ in the do not get finally attached, the effect is called -E-effect

The distinction between inductive and electromeric effect:

3. Resonance
Resonance Definition:
Various Lewis structures, which differ in the positions of non-bonding or ;r -electrons but not in the relative positions of atoms, are called resonance structures, contributing structures or canonical forms. This concept is known as resonance
There are some molecules or ions which cannot be represented adequately by a single electronic (Lewis) structure as all the properties of such molecules or ions do not correspond to a single Lewis structure. In such cases, it becomes necessary to represent the molecule or ion by writing two or more Lewis structures which differ in the arrangements of valence electrons but the basic structure involving cr -bonds remains the same.
It should be remembered that resonance is not a phenomenon, because there is no real existence of different resonance structures. These structures are all imaginary and are taken into consideration to explain the different physical and chemical properties of molecules or ions. The actual structure of the molecule or the ion lies in between these structures. We say that the actual molecule or ion is a resonance hybrid (weighted average) of all these resonance structures.
Resonance is also known as mesomerism. The various resonance structures are connected by double-headed arrows. They contribute to the actual structure in proportion to their stability. The magnitude of internal energy of the resonance hybrid of a molecule or ion is less than that of any resonating structure. Thus the molecule or ion gets stabilised by resonance
NCERT Solutions Class 11 Chemistry Organic Reaction Mechanism
Examples:
1. Benzene molecule can be represented as a resonance ~ hybrid (III) of two Kekule structures:
I and II. Neither ofthe two structures can fully explain all the properties of benzene. For example, both structures I and II contain two types of carbon-carbon bonds such as C—C (1.54 Å) and C = C (1.34 Å). But actually, it has been found that all the 6 carbon-carbon bonds in benzene are of equal length (1.39 Å).
This suggests that the actual structure of benzene can neither be represented by I nor by II but by a resonance hybrid of these two structures in which all the six carbon-carbon bonds are of equal length and lie in between carbon-carbon single bond length of 1.54 Å and carbon-carbon double bond length of 1.34 Å. So, benzene is quite often represented by the non-Lewis structure III. The circle inside the ring indicates completely delocalised 6 n -electrons. Since I and II are exactly equivalent, They are of the same stability and make equal contributions to the hybrid

2. Carbonate ion (CO32-) may be represented as a resonance hybrid ofthe following three structures:
IV, V and VI. None of the three structures can individually explain all the properties of carbonate ions. For example, in all three structures, the carbon-oxygen single bond (1.43Å) and carbon-oxygen double bond (1.20 Å) are present.
However, it has been found experimentally that all the carbon-oxygen bonds in carbonate ion are equal in length (1.28 A) and this bond length is slightly greater than that of the double bond but less than that of the single bond. All the three carbon-oxygen bonds are equivalent. So, the structural formula of the carbonate ion denotes a state equidistant from the three structures IV, V and VI and it is frequently expressed by the non-Lewis structure VII.

Rules for writing meaningful resonance structures:
The following rules are to be followed while writing realistic resonance structures:
- The various resonance structures should differ only in the positions of electrons and not in the positions of atoms, Le., the basic structure involving cr -bonds should remain undisturbed.
- The number of paired and unpaired electrons in each resonance structure must be the same.
- All the atoms involved in the process of resonance must be coplanar (or nearly coplanar).
- All the resonance structures should have nearly the same energy.
- Each resonance structure must be a bona fide Lewis structure, i.e., all atoms in a resonance structure must exhibit proper valencies.
- For example: There must not be any structure with pentavalent carbon, pentavalent nitrogen, bivalent hydrogen and so on.
Resonance energy:
The difference in internal energy between the actual molecule (observed value) and that of the resonance structure having the lowest internal energy or highest stability (obtained by calculation) is called resonance energy. The resonance energy is greater when
- The contributing structures are all equivalent and
- The number of contributing structures of roughly comparable energy is greater.
Calculation of resonance energy:
Resonance energy is not a measurable quantity. It can only be estimated from thermochemical data. If the theoretically calculated internal energy of a gram-mole of the most stable resonance structure is EC and the experimentally determined internal energy of the actual molecule (resonance hybrid) is EO, then the resonance energy ER = EC-EO. Resonance energy is expressed in kcal mol-1 or kj mol-1.
The greater the resonance energy, more the stability of the compound. The resonance energy becomes maximum when the contributing structures are equivalent, i.e., have equal energy content. Also, the more the number of resonance structures having a large contribution, the greater will be the resonance energy.
In determining the relative stabilities of similar molecules or ions, resonance energy is an important factor among various other factors like bond energy, internal strain etc.
Example:
The resonance energy of benzene can be calculated from the heat of hydrogenation values. The heat of hydrogenation is the quantity of heat evolved when the mol of an unsaturated compound is hydrogenated. Cyclohexene containing 1 double bond has a heat of hydrogenation of 28.6 kcal mol-1.
We might reasonably expect 1,3,5-cyclohexatriene to have a heat of hydrogenation of about three times as large as cyclohexene, i.e.,3 × 28.6 = 05.8 kcal .mol-1.
The value for benzene is 49.8 kcal .mol-1. It is 36 kcal .mol-1 less than the expected value. So, benzene evolves 36 kcal less energy per mole than predicted. This can only mean that benzene is more stable than hypothetical cyclohexadiene by 36 kcal .mol-1 energy. This 36 kcal. mol-1 energy is the resonance energy of benzene.

Fundamental Concepts of Organic Reaction Mechanism Class 11 Notes
Relative contributions of resonance structures towards resonance hybrid:
All resonance structures do not contribute equally towards resonance hybrid. Relative contributions of resonance structures towards resonance hybrid depend on their relative stabilities. The more stable the resonance structure, the more will be its contribution to resonance hybrid.
Factors that govern the stability of a resonance structure and its relative contribution towards hybrid are :
Rule 1:
Non-polar resonance structures, being more stable than the dipolar resonance structures, contribute more towards the resonance hybrid.
Example:
In the following alkadiene, the first resonance structure is more stable and thus contributes more than the second dipolar resonance structure

Rule 2:
Resonance structures with a greater number of covalent bonds are more stable and contribute more towards the resonance hybrid.
Example:
In the following acyl cation, the second resonance structure having a greater number of covalent bonds is more stable and more contributing.

Rule 3:
In case of anions, the most stable structure is the one in which the negative charge resides on the most electronegative the one in which the positive charge resides on the least electronegative atom. So, these structures are more contributing.
Example:
Resonance structures II and IV of the following anion and cation are relatively more stable hence more contributing towards their respective resonance hybrids.

Rule 4:
Canonical structures in which octets of all the atoms are fulfilled are relatively more stable and therefore, make a larger contribution towards the resonance hybrid.
Example:
The second resonance structure of the following acylium ion is more stable and more contributing because all the atoms have octets of electrons in their valence shells (except H which has a duplet).

Rule 5:
Aromatic resonance structures are more stable and more contributing than the non-aromatic resonance structures having the same number of covalent bonds. Example: The aromatic resonance structure I of benzyl cation is more stable and more contributing than the non-aromatic resonance structure II.
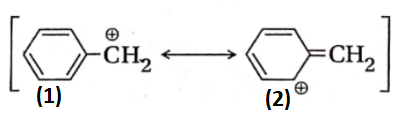
Organic Reaction Mechanism Overview Class 11 Chemistry
Rule 6:
A resonance structure having two units of charge on the same atom is not stable and hence it has a very poor contribution. Again, structures having like charges on adjacent atoms are highly unstable and hence it has a negligible contribution. On the other hand, a resonance structure having two dissimilar charges close to each other is relatively more stable and more contributing than the structure in which the charges are further apart.

Rule 7:
The resonance energy of a system involving monopolar resonance structures is greater than that involving dipolar resonance structures. So the former type of systems (i.e., molecules or ions) are more stable than the latter type.
Example:
Carboxylate ion is more stable (in fact more stabilised by resonance) than the corresponding carboxylic acid

Some facts about resonance structures:
- Resonance structures are not real.
- Resonance structures are not in equilibrium with each other.
- Resonance structures are not isomers because the two isomers differ in the arrangement of both atoms and electrons, whereas resonance structures differ only in the arrangement ofelectrons
Effect of resonance on the properties of molecules:
The following properties of different molecules or ions can be explained by resonance
1. Bond length
Because of resonance, the single bond present in a molecule or ion may acquire a partial double bond character with a consequent decrease in bond length. Similarly, the double bond may acquire some single bond character with a consequent increase in bond length.
Example: Due to resonance, the C — Cl bond (1.69 Å) of vinyl chloride (CH2 = CHCl) becomes shorter than C — Cl bond (1.76 Å) of ethyl chloride and its C=C bond (1.38 Å) becomes longer than the C=C bond (1.34 Å) of ethene

2. Dipole moment
As a result of resonance, both the magnitude ofthe charge separated (e) and the distance between two charged centres (d) in any molecule may increase. So, the value of dipole moment (p = e × d) increases.
Example:
Due to resonance, the amount of charge separated and the distance between the centres of charges in nitroethane (CH2=CHNO2) is greater as compared to nitroethane (CH3CH2NO2). Consequently, the dipole moment <p) nitroethane is greater than that of nitroethane.

3. Acidity And Basicity Of Organic Compounds
Acidic character of phenol:
The greater the ease with which a compound releases proton (H+) in its aqueous solution, the stronger it will be as an acid. That phenol is acidic and is a stronger acid than alcohol can be well explained in terms of resonance. Phenol in its aqueous solution ionises to produce phenoxide ions as follows

CBSE Class 11 Organic Reaction Mechanism Fundamental Concepts
Phenol can be represented as a resonance hybrid of the following resonance structures (I -V):

Due to the contribution ofthe resonance structures n, in an IV the O -atom becomes positively charged. Because of this, the polarity of the O—H bond increases and hence the tendency of O—H bond fission (to release a proton) also increases. On the other hand, no such resonance is possible in a molecule of alcohol.
So, the alcoholic O — H bond is relatively less polar and the tendency of bond cleavage resulting in proton release is indeed very small. Hence, phenol is more acidic than alcohol.
Alternative explanation:
This relative acidity may also be explained by considering the phenol-phenoxide ion and Alcohol-alkoxide ion equilibria. Like phenol, phenoxide ion may also be represented as a resonance hybrid of the following (VI – X) resonance structures:
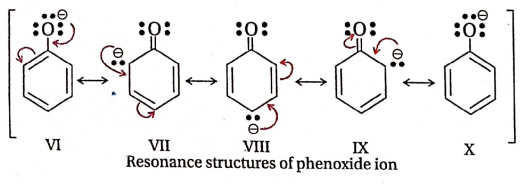
Three (II, III and IV) out of five resonance structures of phenol involve charge separation, but the resonance structures of phenoxide ion involve no charge separation. The negative charge is only delocalised. Because of this, phenoxide ion is more resonance stabilised than phenol.
As a consequence, the equilibrium of phenol-phenoxide ion tends to shift towards the right, i.e., phenol exhibits acidic properties by releasing proton (H+) easily. On the other hand, both alcohol and alkoxide ions can be satisfactorily represented by single (localised) structures. Due to the absence of differential stabilisation caused by resonance, alcohol is very reluctant to produce alkoxide ions. So, phenol is a stronger acid than alcohol.
The basic character of aniline:
Aniline is a weak base and its basicity is much weaker than aliphatic amines (RNH2). This can be explained by resonance. It can be represented as a resonance hybrid ofthe following resonance structures:

1. An unshared pair of electrons on the N-atom of aniline becomes involved in resonance interaction with the ring. As a result, N-atom acquires a partial positive charge. Consequently, aniline exhibits little tendency to take up a proton. So aniline is a weak bases
2. However, in the case of aliphatic amines, similar delocalisation of electrons by resonance is not possible, Naturally the electron density on N-atom is not reduced. In fact, due to the +1 effect of the alkyl group (R-), the electron density on Natom is somewhat increased.
As a consequence, nitrogen can easily donate its electron pair to a proton (H+) to combine with it. Thus, aniline (C6H5NH2) is a weaker base than aliphatic amines (RNH2). Apart from this, relative basicity can also be explained by considering aniline-anilinium ion and amine ammonium ion equilibrium systems.
Aniline is a resonance hybrid of five resonance structures (I-V).In the conjugate acid anilinium ion, the lone pair of electrons on the N atom is localised in the N —H bond and so, only two structures (VI and VII) can be drawn for its hybrid. Therefore, aniline is more resonance-stabilised concerning the anilinium ion. As a result of this, protonation of aniline is disfavoured.
None of the aliphatic amine and its conjugate acid can be stabilised by resonance. The conjugate acid is stabilised by the weak +1 effect ofthe -R group. Protonation ofthe aliphatic amine is, therefore, not disfavoured and is is somewhat favoured. Thus, aromatic amines are weaker bases than aliphatic amines.

Finally, in aromatic amine, the amino group is attached to sp² -carbon (more electronegative), whereas in aliphatic amine, it is attached to sp³ -carbon (less electronegative). This factor is also partly responsible for decreased basicity of aromatic amines than aliphatic amines.
Resonance effect or mesomeric effect
Resonance effect or mesomeric effect Definition:
The displacement of non-bonding or electrons from one part of a conjugated system (having alternate single and double bonds) to the other part causing permanent polarity in the system (creating centres of high and low electron density) is called resonance effect (R-effect) or mesomeric effect (M-effect)
Organic Reaction Mechanism NCERT Class 11 Notes
There are two types of resonance or mesomeric effect:
1. +R or +M-effect:
An atom or a group is said to have a +R or +M effect if it involves the transfer of electrons away from the atom or the substituent group attached to a double bond or a conjugated system
+ R or +M groups: —OH, —OR, —SH , — NH2, —Cl, — Br, —I etc.
Example: +M effect of Cl -atom in vinyl chloride may be shown as follows

2. — R or —M- effect:
An atom or a group is said to have a — R or — M effect if it involves the transfer of electrons towards the atom or the substituent group attached to a double bond or a conjugated system.
R or -M groups: >C= O, —CHO, —COOR, — CN, —NO2 etc.
Example: -M -effect of —CHO group in acetaldehyde may be shown as follows:

4. Hyperconjugation
Hyperconjugation Definition:
When a carbon containing at least one H -atom is attached to multiple bonds such as C=C, C ≡ C, C= O, C ≡ N etc., the cr -electrons of the C — H bond become involved in delocalisation with the π electrons of the unsaturated system, i.e., there occurs a σ-π conjugation.
Similarly, σ-p type of conjugation may also take place when a carbon-containing at least one H-atom is attached to a carbon-containing partially filled or vacant p -p-orbital. This special type of resonance or conjugation, giving stability to the species (molecule, free radical or carbocation) is called hyperconjugation.
Hyperconjugation causes a permanent polarity in the molecule and is known as the hyperconjugation effect
Examples:
1. Incaseofpropene (CH3CH=CH2)

Although one C—H bond ofthe methyl group is shown to be broken in each hyperconjugative structure, H+ is never free from the rest of the molecule nor does it change its position in the molecule. However, from the point of view of apparent fission of the C —H bond, hyperconjugation is also called no-bond resonance.
2. In the case of ethyl cation, hyperconjugation may be shown as:
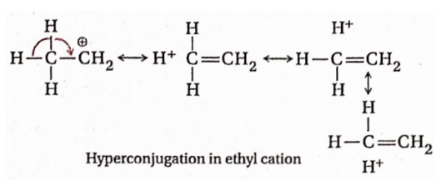
3. In Case of ethyl radical:

Conditions for effective hyperconjugation:
For effective hyperconjugation, the p-orbital concerned and the a-C —H bond, i.e., the sp³-s orbital must remain in the same plane. Orbital representations of hyperconjugation in propene and ethyl cation are shown as follows:

Although the stability of a molecule, ion or free radical increases due to hyperconjugation, this stability is less than that contributed by resonance. After the names of the scientists who proposed this theory, hyperconjugation is also called the BakerNathan effect.
Effects of hyperconjugation:
1. Relative stabilities of alkenes:
The stabilities of alkenes can easily be explained by hyperconjugation. The greater the number of a -hydrogen atoms (II-atom present on the carbon atom attached directly to a double bonded carbon), i.e., the greater the number of hyper conjugative structures, the higher the stability of the alkene due to hyperconjugation
Example:
2-Methylpropcne [(CH)2C=CH2 having 6 hyperconjugable or-H atoms gives 6 no-bond resonance structures while the isomeric compound, 1 -butene (CH3CH2CH = CH2) having only 2 hyperconjugable a-H atoms gives only 2 no-bond resonance structures.
It thus follows that 2-methyl propyne is thermodynamically more stable than its isomer
Directive influence of alkyl groups:
The ortho- and paradirective influence of alkyl groups can be explained by hyperconjugation.
Example:
Directive influence of the —CH3 group in toluene, can be explained based on hyperconjugation as follows:

As a result of hyperconjugation, the electron density at ortho- and para-positions increases and as a consequence, the electrophilic substitution reactions in toluene occur mainly at these two positions. It thus follows that the alkyl groups are o, p-directing.
Class 11 Chemistry Reaction Mechanism Basic Concepts
Relative stabilities of carbocations:
Due to hyperconjugation, the C — H bonding electron pair is attracted towards the positively charged C -atom of the carbocation.
This helps in dispersing the positive charge in different parts of the alkyl group, i.c., charge delocalisation resulting in instability of the carbocation.
As the number of a-H atoms increases, the number of no-bond resonance structures of carbocation increases which enhances the extent of charge delocalisadon and consequent stabilisation.
Hence, the order of stability of ethyl, isopropyl and left-butyl cation, due to hyperconjugation is:

Relative stabilities of free radicals:
Because of hyperconjugation, the odd electron of a free radical undergoes delocalisation for which it becomes stabilised.
With the increase in the number of α-H atoms, the number of no-bond resonance structures of a free radical increases and as a result, delocalization of the odd electron takes place to a greater extent and the stability of free radicals also increases.
Therefore, the stability of ethyl, isopropyl and for-butyl radicals follow the order:

Bond length:
Because of hyperconjugation, the carbon-carbon single bond in propene (CH3—CH=CH2) acquires some double bond character and the carbon-carbon double bond acquires some single bond character.
As a result, the C — C bond in propene is found to be a little shorter (1.488 Å) than the normal C —C bond (1.543 Å) in ethane and the C=C bond is found to be a little longer (1.353 Å) than the normal C=C bond (1.334 Å) in ethylene
Electron-releasing power of alkyl groups attached to unsaturated systems or electron-deficient carbon atoms:
This depends on the number of or-H atoms. The methyl ( —CH3) group having three α-H atoms has the highest hyperconjugative effect while this effect is non-existent with the t-butyl group (Me3C— ) having no a-H atom. So, electron releasing power of various alkyl groups when attached to a double bond (or an electron-deficient carbon) follows the order:
CH3 →CH3CH3→ (CH3)2CH→ (CH3)3C—
This order is exactly the reverse of the order of the +I-effect of these alkyl groups
5. Steric hindrance or steric strain
Steric hindrance Definition:
Steric hindrance or steric strain refers to repulsive interactions between non-bonded atoms or groups which arise when the atoms or groups come very close to each other.
When two non-bonded atoms in a molecule Are closer to each other than the sum of their van der Waals radii, they repel each other due to spatial -crowding.
The repulsion arises primarily due to electron-electron repulsive forces involving the non-bonded atoms. Such repulsive interactions between non-bonded atoms is known as steric hindrance or steric strain. Steric strain is responsible for decreased stability or destabilisation of molecules.
When the sheer bulk of groups at or near a reacting site of a molecule hinders or retards a reaction, it is called steric hindrance. On the other hand, if the constituent atoms or groups of a molecule or ion owing to their bulky nature require more space than what is available for them, i.e., when they are forced too close to one another, then mechanical interference forced too close to one another, mechanical interference ion is then said to be under steric strain. Steric strain makes the species unstable, i.e., its energy increases
Example:
In cis. 2-butene, two -CH3 groups lying on the same side of the double bond are quite close to each other and so they get involved in steric interaction. In trans-2- butene, two —CH3 groups lying on the opposite sides of the double bond are far apart from each other so they are not involved in steric interaction. Thus, cis-2-butene is thermodynamically less stable than Frans-2-butene

The heat of hydrogenation of cis-isomer is 28.6 kcal mol-1 and for trans-isomer is 27.6 kcal .mol-1. This observation agrees with the relative stabilities of these isomeric alkenes. Because of steric hindrance, tertbutyl chloride does not hydrolysis by an SN2 mechanism

Effect on Stability or reactivity:
Steric hindrance is responsible for decreasing the stability and increasing the reactivity of many compounds. Due to steric strain, resonance or delocalisation of electrons may be inhibited (Steric inhibition of resonance or SIR). Again, steric hindrance created at the reaction centre decreases the rate of that reaction or does not even allow the reaction to occur. So, steric hindrance plays a vital role in determining the reactivity of a compound
The acidic character of substituted aromatic acids, phenols and basic character of substituted aromatic amines:



NCERT Class 11 Chemistry Organic Reaction Mechanism Summary

