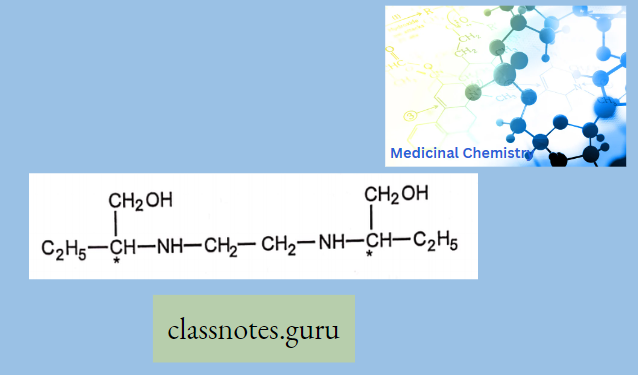
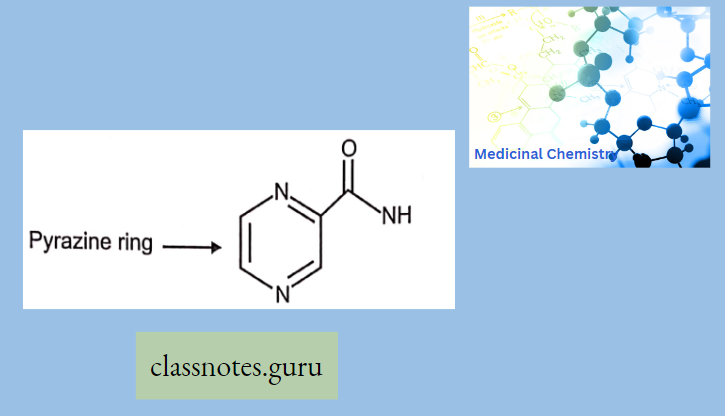
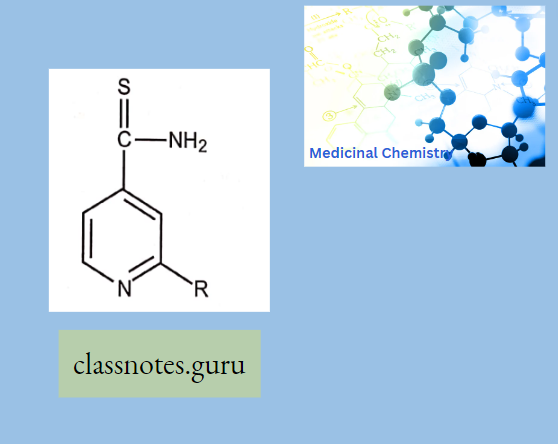
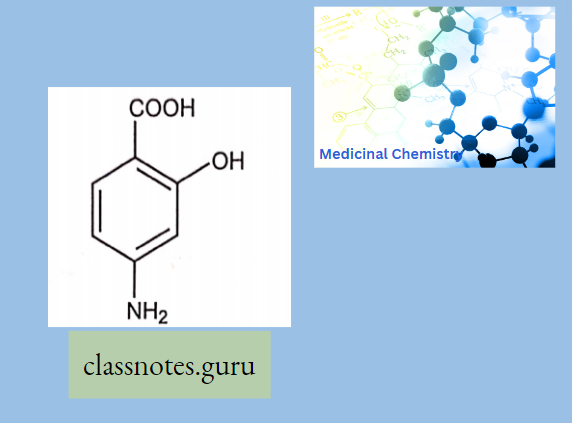
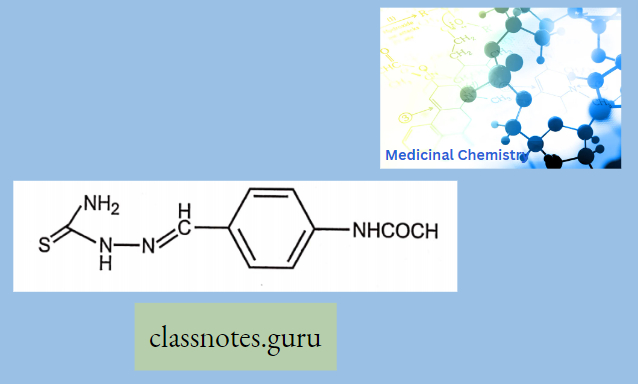
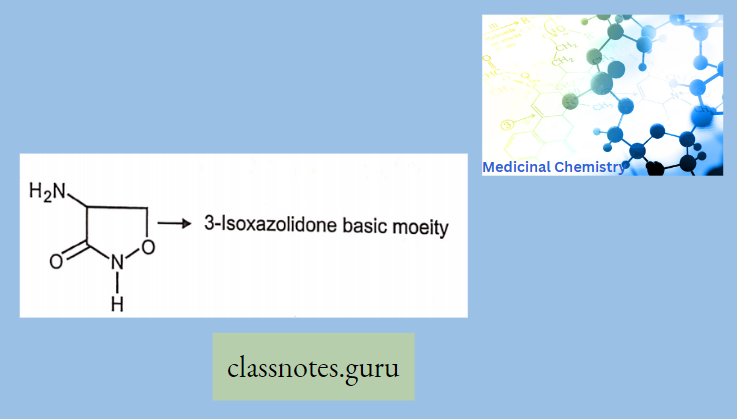
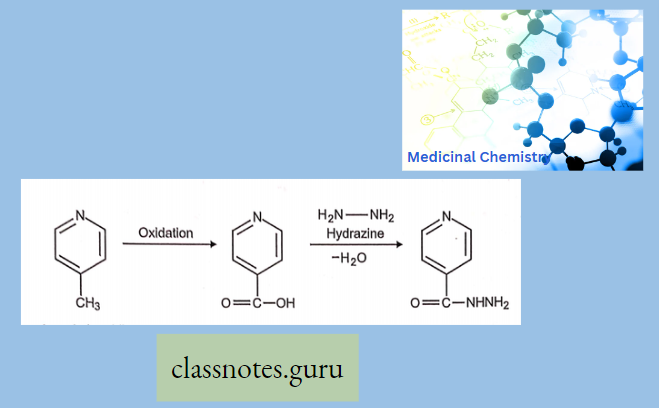
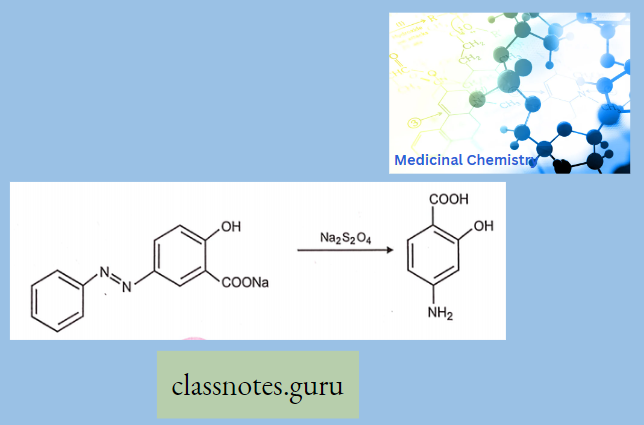
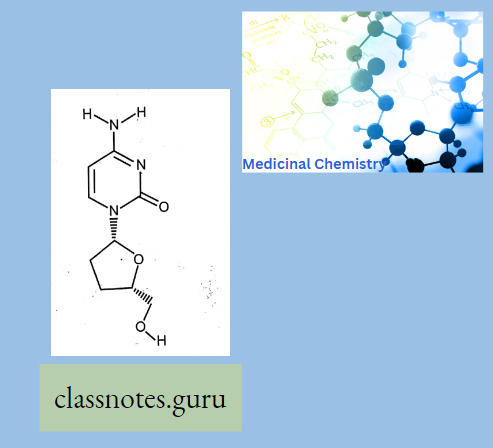
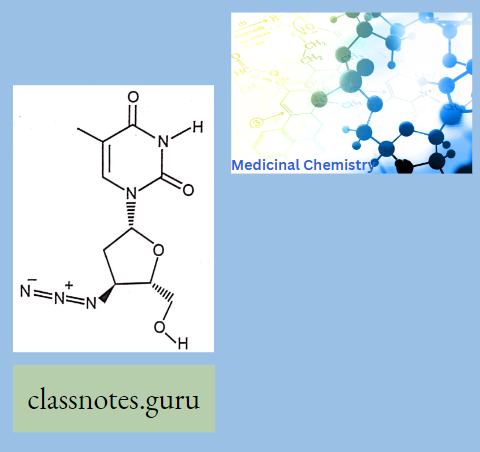
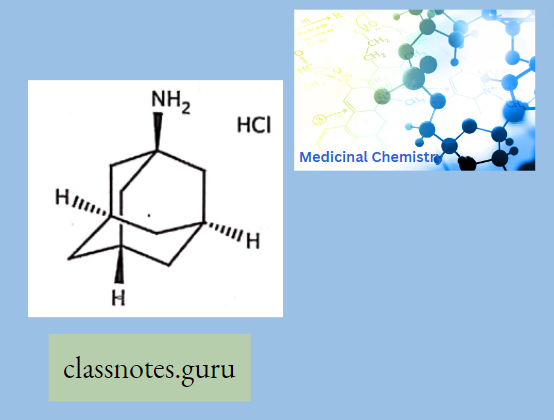
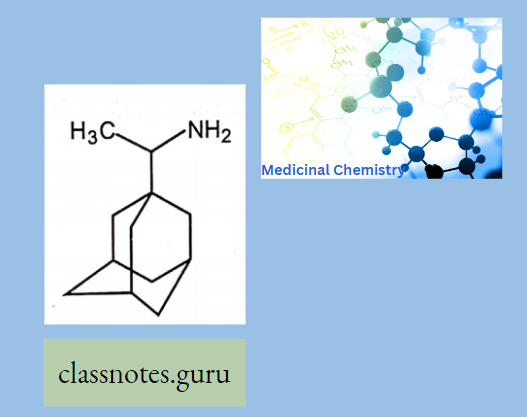
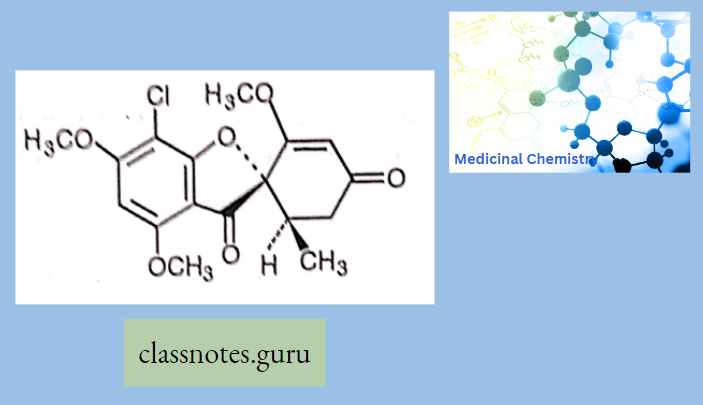
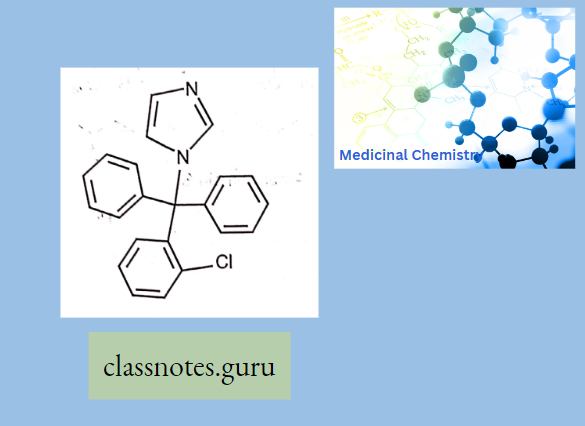
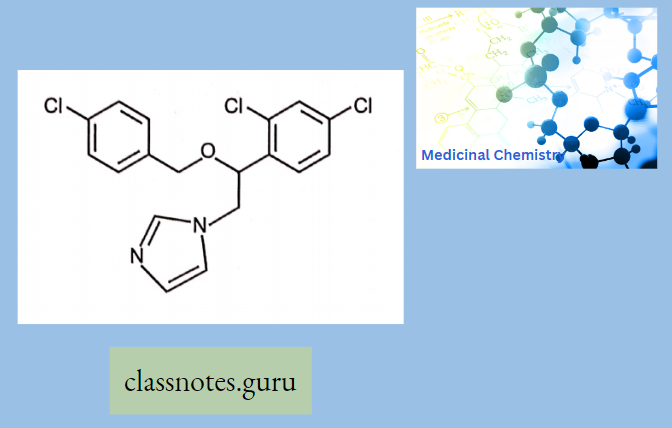
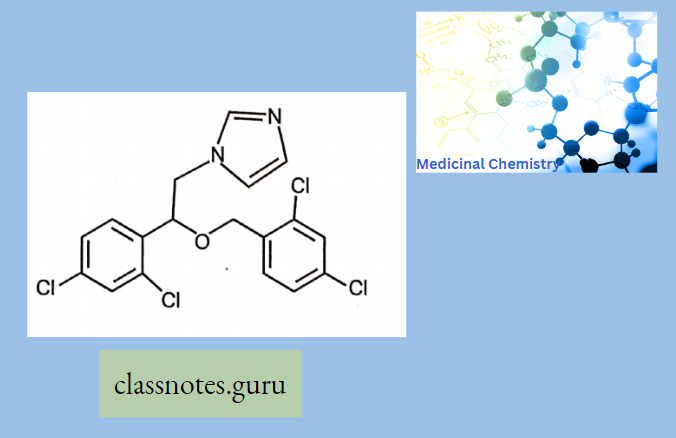
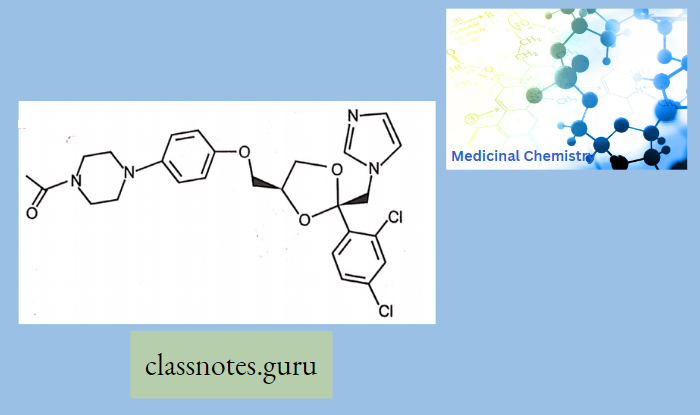
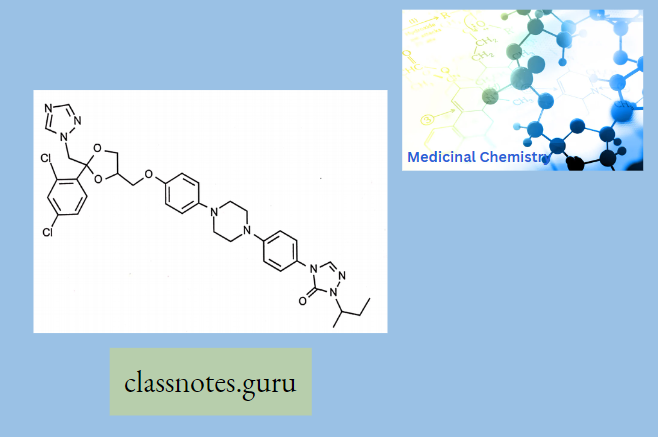
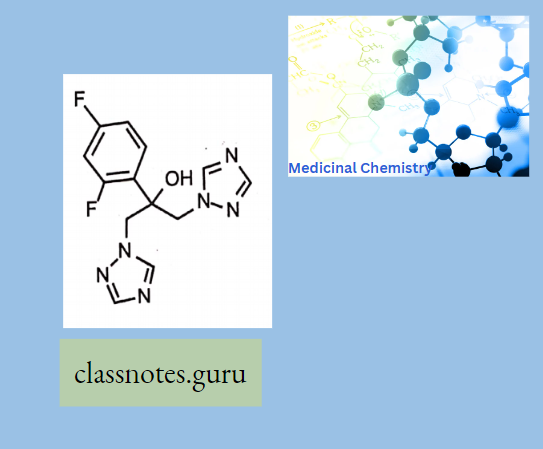
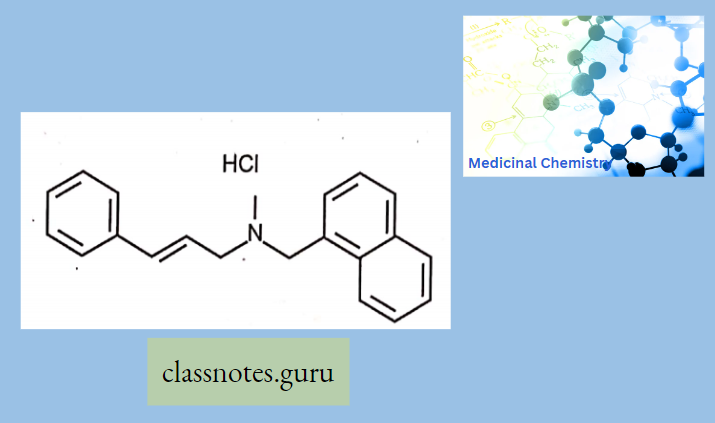
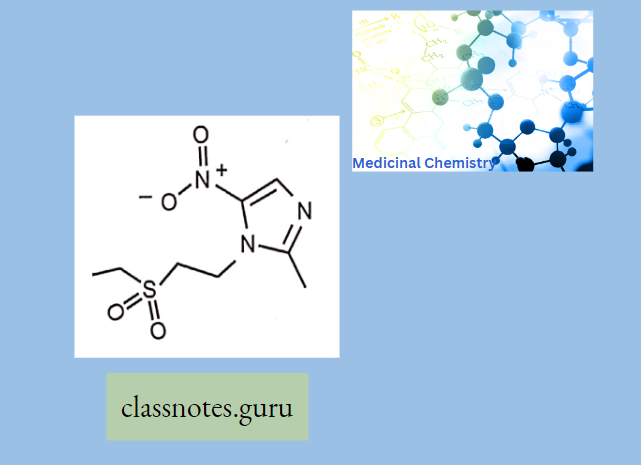
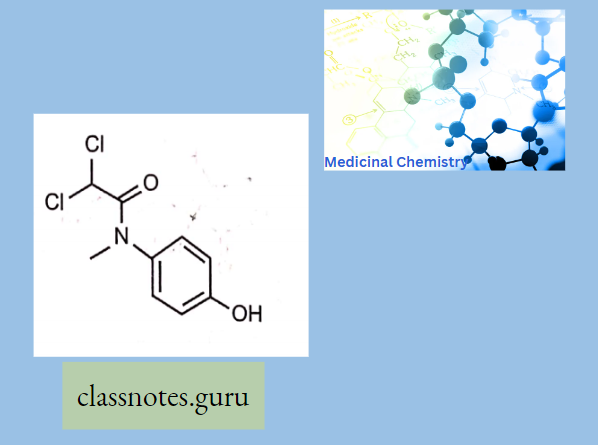
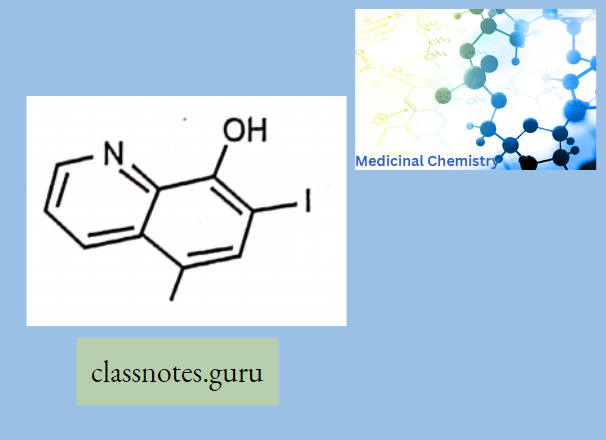
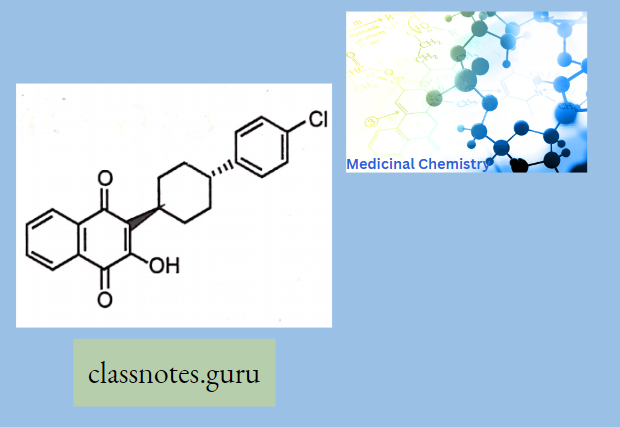
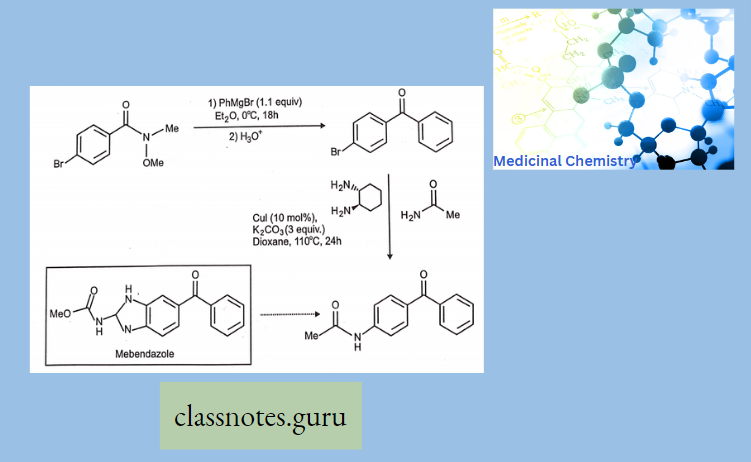
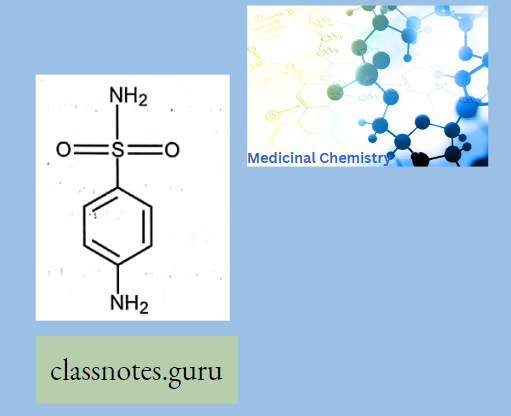
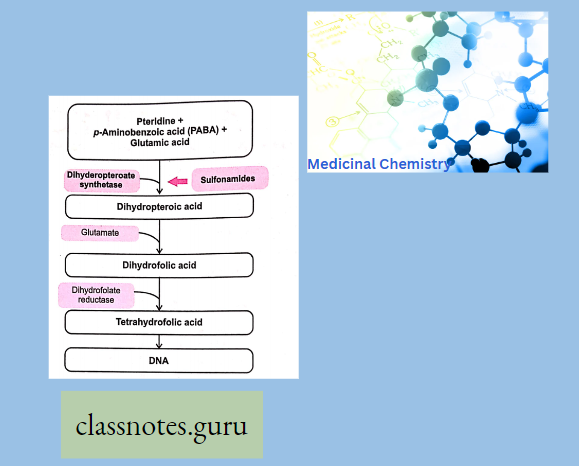
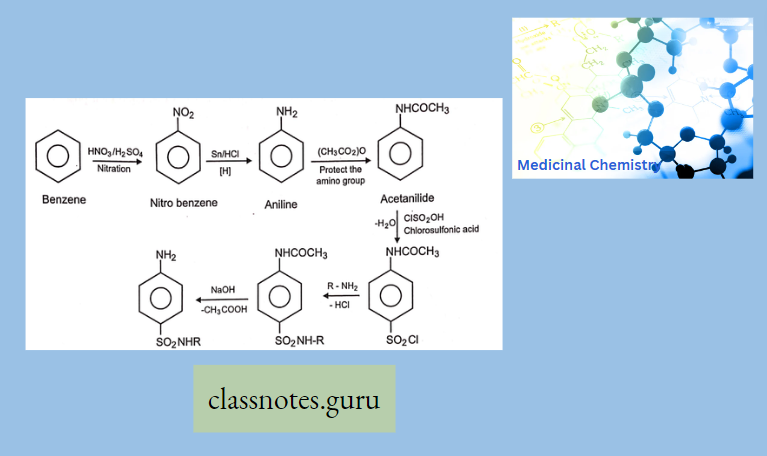
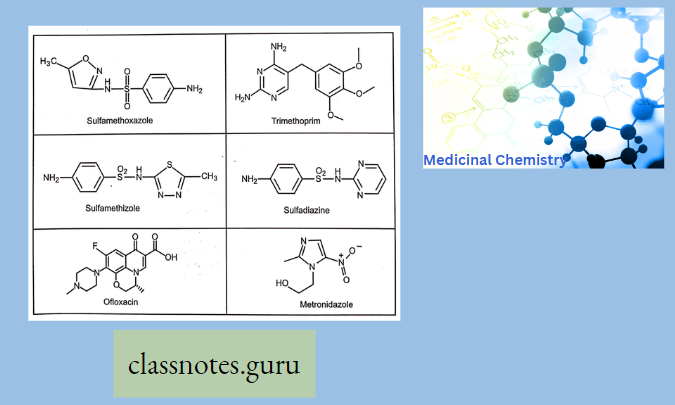
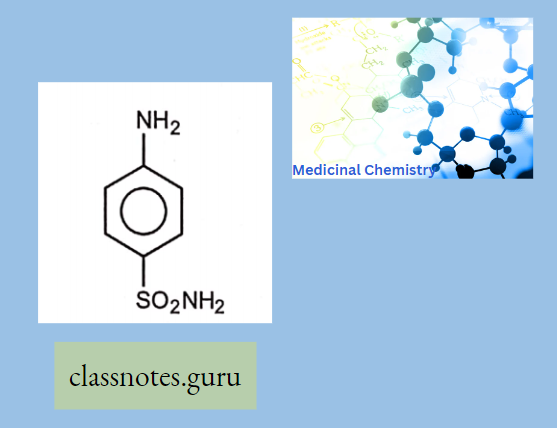
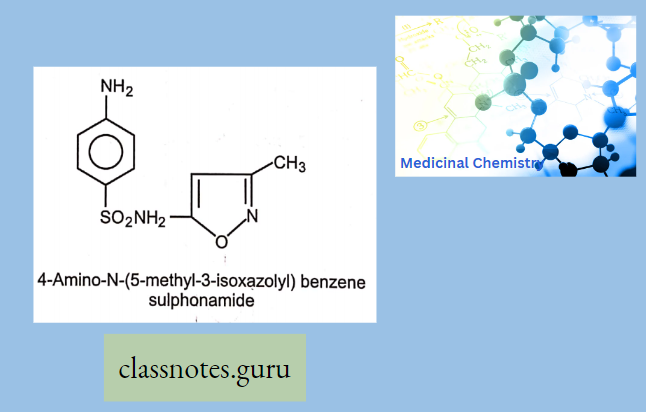
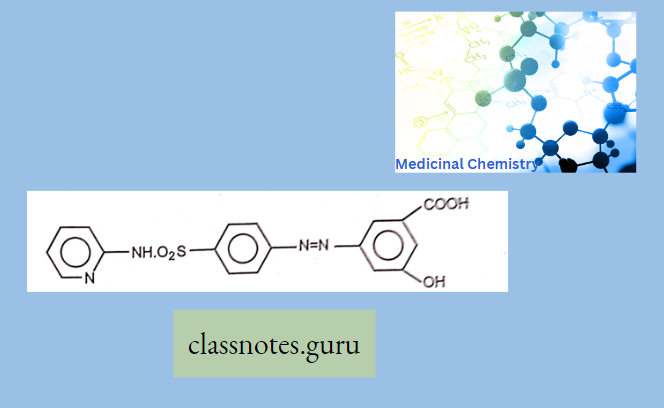
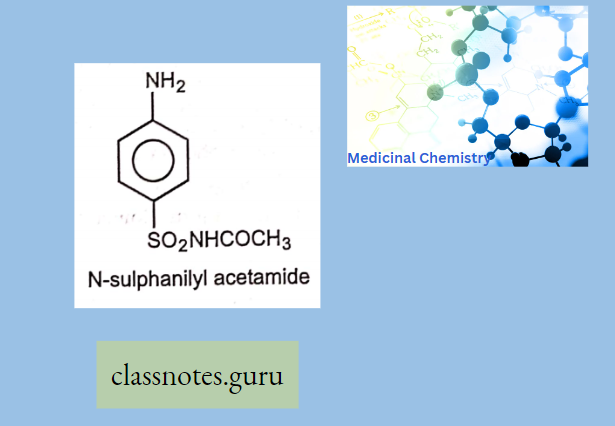
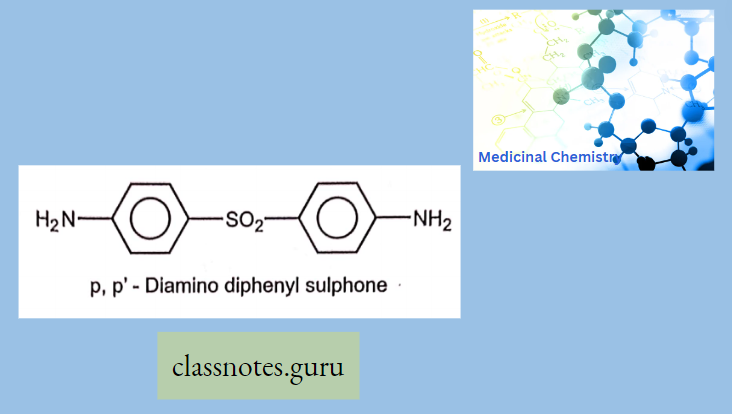
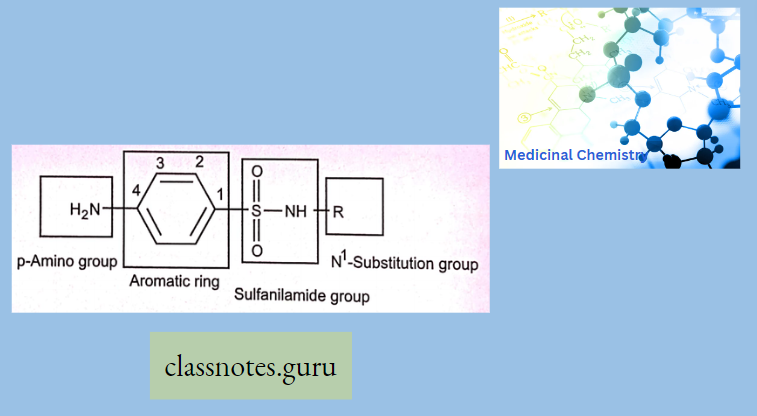
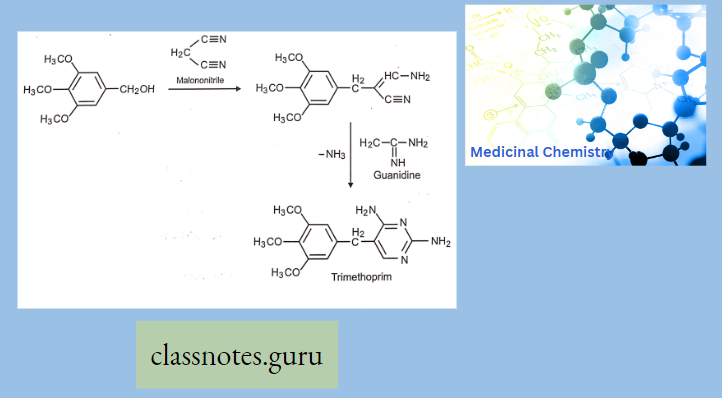
Antibiotics Definition
A substance produced by one microorganism that selectively kills or inhibits the growth of another.
- Antibiotic: A class of substances that can kill or inhibit the growth of some groups of microorganisms. Originally antibiotics were derived from natural sources (for example penicillin from molds), but many currently used antibiotics are semi-synthetic and modified with additions of manmade chemical components.
- Antimicrobial: In this document, the term “antimicrobial” is used inclusively to refer to any agent (including an antibiotic) used to kill or inhibit the growth of microorganisms (bacteria, viruses, fungi, or parasites). This term applies whether the agent is intended for human, veterinary, or agricultural applications.
History Of Antibiotic Development
- 1928: Alexander Fleming noted that the growth of bacterial colonies is inhibited by the coexistence of fungal colonies. Fleming concluded that the material produced by the fungus is not worth being used clinically because it is difficult to isolate.
- 1930: Florey et al isolated a compound by freeze-drying from the fungus and named it penicillin which has an antibiotic effect.
- 1945: D. Hodgkins illustrated the chemical structure of penicillin and gave the excuse for Fleming’s failure in isolating penicillin (Why?).
- 1957: Sheehan develops a synthetic route for the production of penicillin.
- 1958: Beechams isolates 6-aminopenicillins acid (6-APA) to be used as an intermediate for semi-synthetic penicillin derivatives.
- The careless use of penicillin led to the emergence of bacterial resistance.
- In 1976, Beechams isolated a natural product called clavulanic acid that is effective in preventing enzymatic digestion of penicillin in resistant strains of bacteria.
Read and Learn More Medicinal Chemistry III Notes
Bacterial Cell-Wall:
- Bacteria have cell walls to survive a large range of environmental conditions, such as varying pH, temperature, and osmotic pressure.
- Human and animal cells have no cell wall, which makes it a perfect target for internally- used antibiotics.
- The structure of the wall consists of a parallel series of sugar backbones containing two types of sugar [ N -acetylmuramic acid (NAM) and N – acetyl glucosamine (NAG)].
Batalactum Antibiotics
The p-lactam antibiotics are a large class of diverse compounds used clinically in both the oral and parenteral forms. The p-lactam antibiotic agents have become the most widely used therapeutic class of antimicrobials because of their broad antibacterial spectrum and excellent safety profile.
Biosynthesis Of Penicillin: It is synthesized within the penicillium by fusing two amino acids (L-cysteine and L- valine)The acyl side chain (R) varies, depending on the components of the fermentation medium.
Synthesis Of Penicillin Analogues
- Fermentation: Addition of different carboxylic acids to fermentation medium to produce penicillin with different acyl side chains – Only suitable for unbranched carboxylic acids – Tedious and time-consuming.
- Complete Synthesis: Long processes and low-yielding (1%)
- Semi-Synthesis: Use a carboxylic acid-deficient fermentation medium to generate 6-aminopenicillanic acid. – 6- APA is (a very weak antibiotic) and reacts with different acyl chlorides to synthesize penicillin analogs.
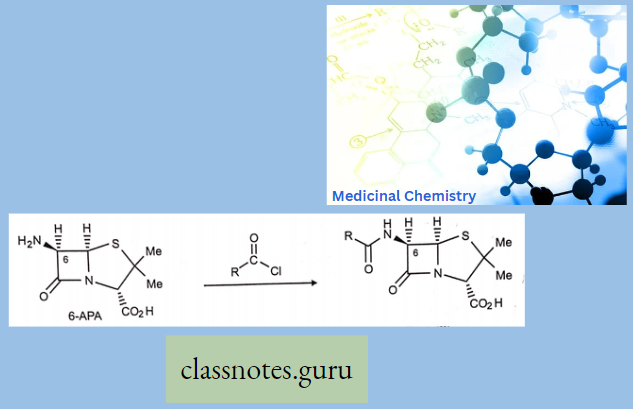
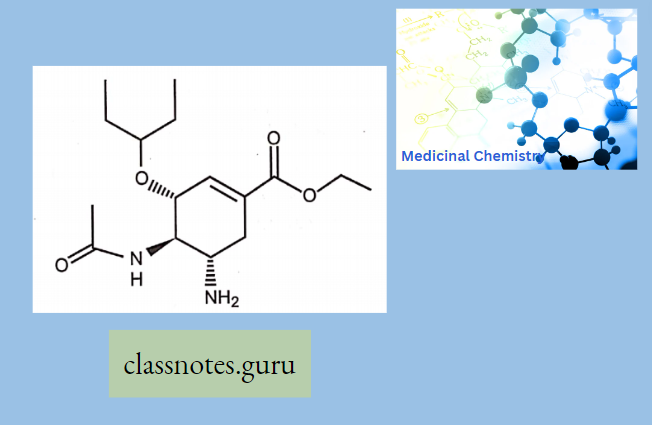
Penicillin: All penicillins are derivatives of 6-aminopenicillin acid (thiazolidine ring is attached to a p—p-lactam ring that carries a secondary amino group (RNH—)) and contains a beta-lactam ring structure that is essential for antibacterial activity.
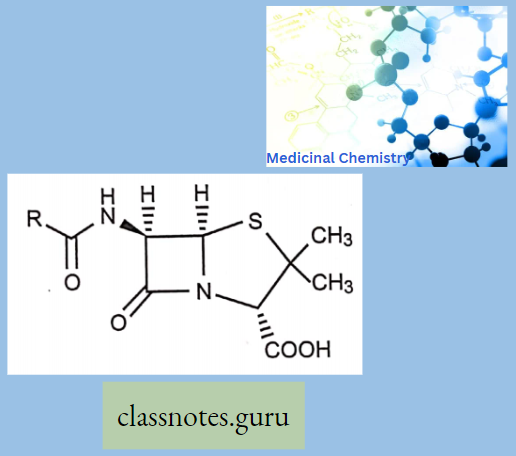
- Beta-lactam antibiotics are narrow-spectrum and bactericidal drugs. Penicillin is obtained from P. cryssogenum.
- Penicillin is degraded by the acidic pH and amide linkage destruction through the b-lactamase enzyme which is produced by gram-negative bacteria. Beta-lactams act only by multiplying cells.
Mechanism Of Action Of Penicillin
They act as an irreversible inhibitor of the enzyme transpeptidase, an enzyme bacteria use to make their cell walls.
- The final transpeptidation step in the synthesis of the peptidoglycan is facilitated by transpeptidase known as penicillin-binding proteins (PBPs).
- PBPs bind to the D-Ala-D-Ala at the end of muropeptides, the peptidoglycan precursors to crosslink the peptidoglycan.
- p-lactam antibiotics mimic the site and competitively inhibit PBP cross-linking of peptidoglycan.
Penicillin Classification
1. Naturalpenicillin’s:
- Benzylpenicillin (penicillin G)
- Phenoxy methyl penicillin (penicillin V)
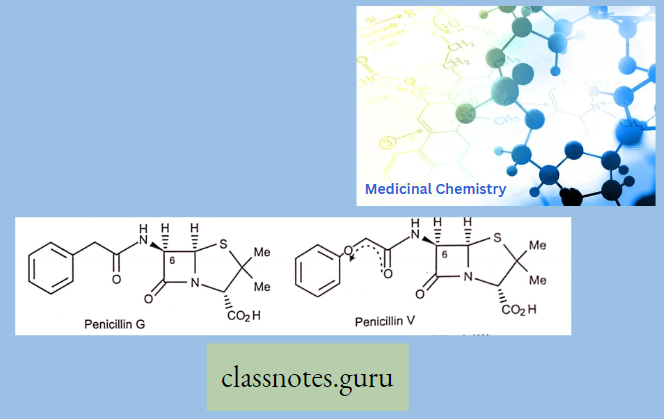
Effective Against: Gram-positive + Less effective against Gram-negative bacteria.
Treatment For Naturalpenicillin’s:
- Tonsillitis.
- Anthrax.
- Rheumatic fever.
- Streptococcal skin infections
Characteristics of Natural Penicillin:
- Narrow spectrum.
- Should be given orally.
- Prone to beta-lactamase.
Problems With Penicillin G:
- It is sensitive to stomach acids.
- It is sensitive to (i-lactamases – enzymes which hydrolyze the [Mactam ring.
- It has a limited range of activities.
2. Aminopcnicillin’s
- Ampicillin.
- Amoxicillin.
The Group Has The Following Properties:
- Hydrophilic NH2 group attached to C that is u to C=0 of the acyl side chain
- The acid stability is enhanced due to the electron-withdrawing effect of Nl L
- No bulky groups at acyl side chain 4 more sensitive to [Mactanuso.
- And COOH groups are ionized with poor absorption from the gut.
- The ionizable groups can be masked to form prodrugs with better absorption
- The ci-carbon becomes chiral (activity of D-isomer L-isomer and penicillin G)
–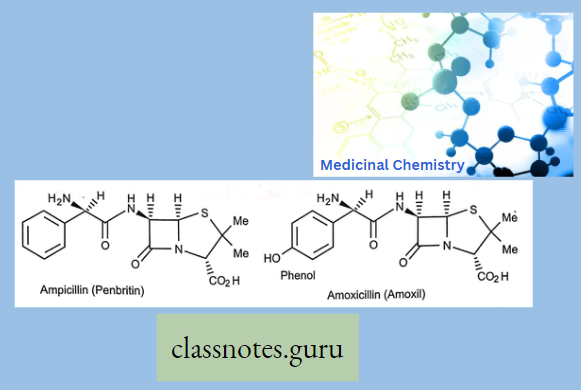
Ampicillin And Amoxicillin Have:
- It has a similar spectrum to Penicillin G but is more active against Gram-ve cocci and enterobacteria.
- Inactive against P. aeruginosa.
- Non-toxic and can be taken orally.
- High doses change gut flora problems such as diarrhea.
3. Broad Spectrum Penicillin’s
Ureidopenicillins:
- Piperacillin
- Mezocillin
- Azlocillin
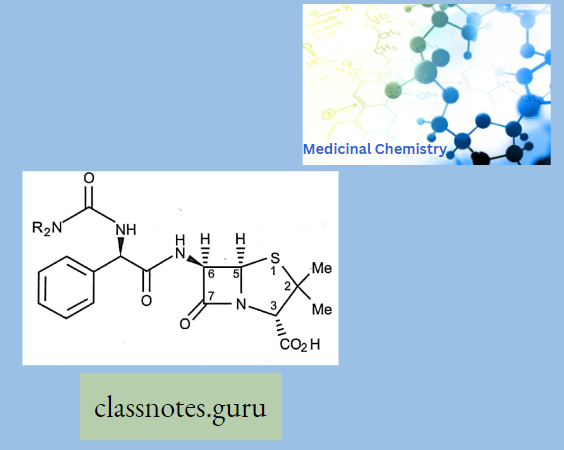
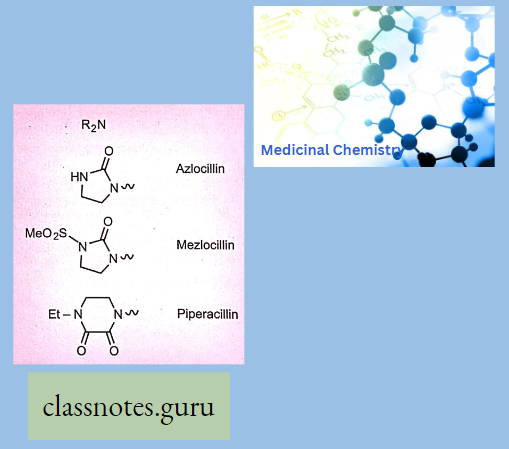
The Group Has The Following Properties:
- Urea functional group attached to C that is a to C=0 of the acyl side chain.
- More active vs. Gram-ve than carbenicillin (more cell-mem permeability).
- Higher activity against P. aeruginosa.
- p-lactamase sensitive.
- Acid-sensitive.
Stereochemistry And Iupac Of Beta – Lactum Ring
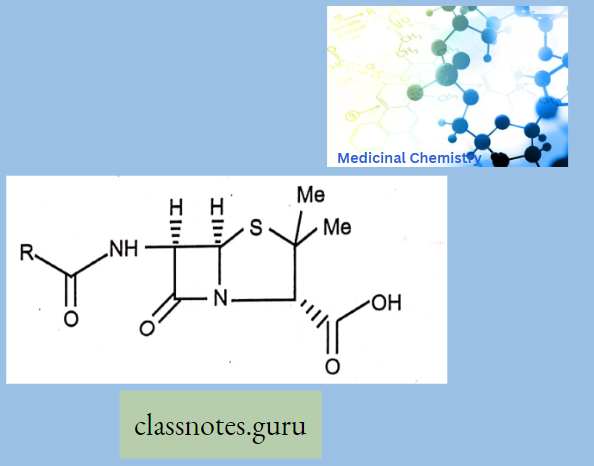
- It has a total of three chiral carbons 3,5 and 6.
- All synthetic and semi-synthetic penicillin have the same absolute configuration (like 3S 5R, 6R).
- Acyl amino and carboxylic acid Trans to each other.
- The lead molecule in the discovery of semi-synthetic penicillin is 6 amino penicillinic acid (6-APA).
- 6-APA is structurally derived from L-valine and L-cysteine.
- All penicillins have a Penam ring as a basic moiety.
- Certain strands of microorganisms destroy p-Lactam antibiotics enzymatically like Penicillanase or p-Lactamase (Open the p-Lactam ring).
Structure-Activity Relationship Of Penicillin:
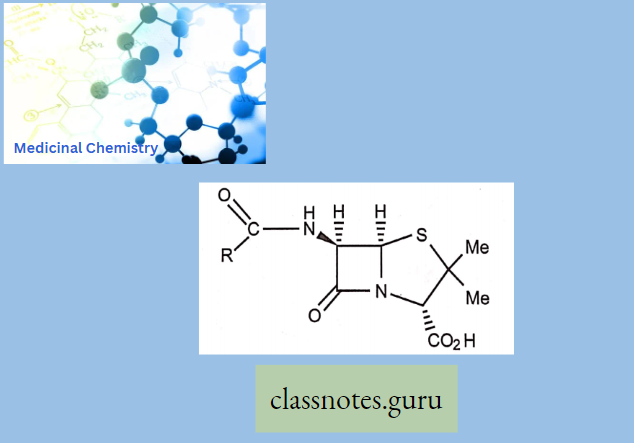
- Amide and carboxylic acid are involved in binding.
- Carboxylic acid binds as the carboxylate ion.
- The mechanism of action involves the p-lactam ring.
- Activity related to p-lactam ring strain (subject to stability factors).
- The bicyclic system increases the p-lactam ring strain.
- Not much variation in structure is possible.
- Variations are limited to the side chain (R).
β-Lactamase Inhibitors
Clavulanic Acid:
- Weak, unimportant antibacterial activity
- Powerful irreversible inhibitor of (β-lactamases) – suicide substrate
- Used as a sentry drug for ampicillin
- Augmentin = ampicillin + clavulanic acid [drug of choice for GIT infection]
- Allows less ampicillin per dose and an increased activity spectrum
- Timentin = ticarcillin + clavulanic acid Penicillanic acid sulfone derivatives
Penicillanic Acid Sulfone Derivatives
Sulbactam
Tazobactam:
- Suicide substrates for (β-lactamase enzymes
- Sulbactam has a broader spectrum of activity as (β-lactamases) than clavulanic acid but is less potent
- Unasyn = ampicillin + sulbactam
- Tazobactam has a broader spectrum of activity as (β-lactamases) than clavulanic acid and has a similar potency
- Tazocin or Zosyn = piperacillin + tazobactam
Some Important Knowledge About Penicillins:
- Penicillin, abbreviated as PCN or pen, is a group of antibiotics that resulted from Penicillium fungi. It is used in the treatment and prevention of diseases caused by Gram-positive organisms, such as syphilis and infections caused by staphylococci and streptococci.
- Several penicillin types counter bacteria to a variety of extents.
- Some of these are flucloxacillin, ampicillin, phenoxymethyl penicillin, and amoxicillin.
- These fungi were originally discovered by a medical student, Ernest Duchesne, in the late 19th Century, and then re-discovered by Alexander Fleming, in 1928, for their antibiotic properties.
- He realized this when a sample of a certain bacteria that he was studying, Staphylococcus, got infected by some mold, and all bacteria cells closest to the mold were perishing.
- With further testing, Fleming learned the mold was creating a bacteria-demolishing substance, which he named penicillin. ,
- The disease-causing bacteria constantly rebuild their cell walls to protect themselves and maintain their structure. Penicillins work by penetrating and destroying these cell walls, consequently killing the bacteria cells.
Cephalosporins
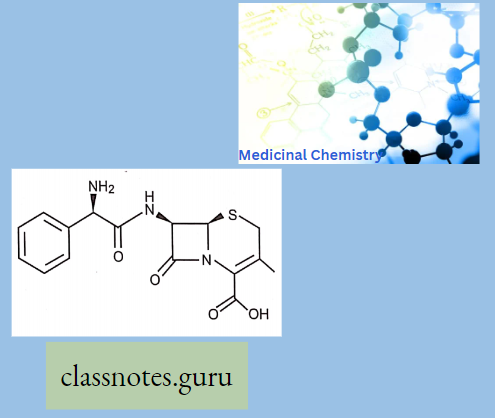
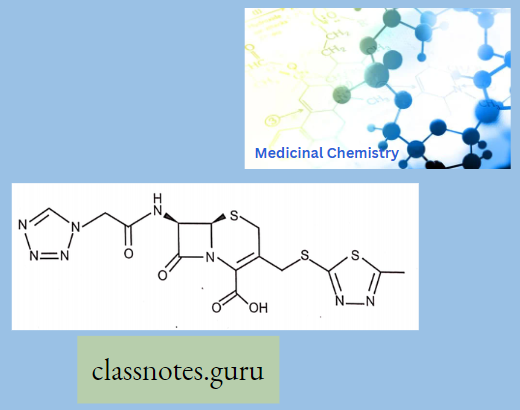
Cephalosporins comprise a large group of semi-synthetic drugs, most of which are derived from cephalosporin C, a substance obtained from a species of Cephalosporium.
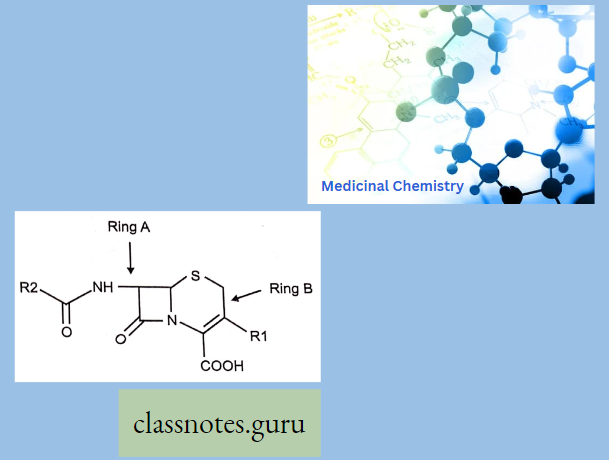
Cephalosporins have a p-lactam ring and a dihydrothiazine ring (7-aminocephalosporanic acid). Additions of any side chain in the p-lactam ring at 7 positions modify the spectrum of activity and the dihydrothiazine ring at 3 positions modify the pharmacokinetic properties.
- Ring A is a four-member p-Lactam ring and Ring B is a six-membered Dihydrothizine ring.
- Cephalosporin having cepham as a basic moiety
- Like 6-APA in penicillin, 7-Amino cephalosporin acid is a lead molecule for the synthesis of semi-synthetic cephalosporin
- Cephalosporin C-True cephalosporin or 7-ACA Cephalosporin P-Acidic antibiotics or Steroidal antibiotic (Fusidin)
- Fusidin- It is a sodium salt of fusidic acid.
- Cephalosporin N-Derivative of 6-APA, Also known as Synnematin N nowadays, is known as Penicillin N.
- All cephalosporins are bactericidal and have the same mechanism of action as penicillin cell wall synthesis inhibition.
- Cephalosporins have greater acid and p-lactamase resistance properties and a wide range of antibacterial activity.
- Most cephalosporins are excreted primarily by renal tubular secretions probenecid inhibits tubular secretion like penicillins.
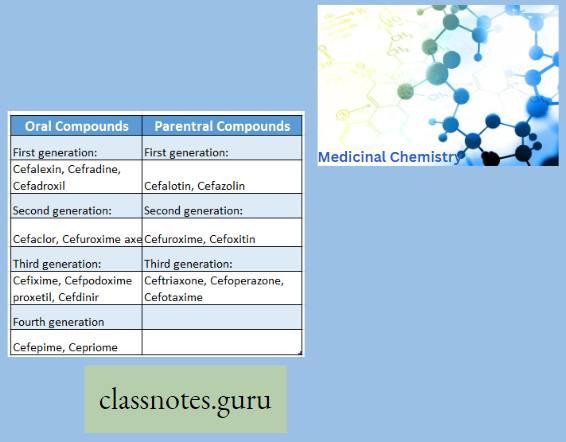
Cephalosporin Classification
Structure-Activity Relationship Of Cephalosporins
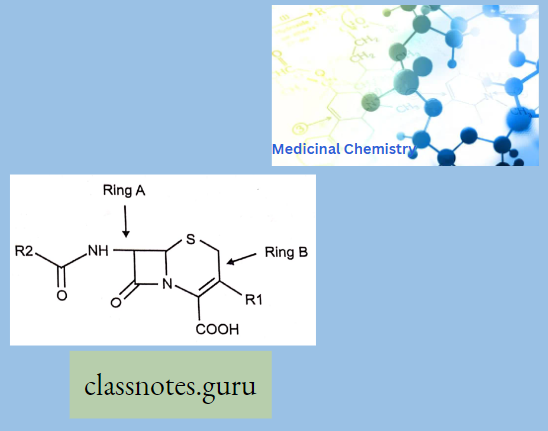
1. Beta-Lactam Ring:
- Required for PBP reactivity and antibacterial activity
- Reactivity is reduced compared to the penicillins (2 reasons)
- Compare mechanism of action, resistance, pharmacodynamics, etc to penicillins
2. 2-Carboxyl Group:
- Acidic: Salt formation, product formulation Prodrug formation Elimination
- profile: Renal
3. X-Substituent:
- Cephalosporins and cephamycins
- Determines, in part, resistance to beta-lactamase inactivation
4. 3- Substituent (R3):
- Chemical or acid stability or instability or Metabolic stability or instability
- Minimal impact on antibacterial activity
- Protein binding and half-life: Heterocycles
- Adverse Reaction and Drug Interaction
5. 7- Substituent (R7):
- Incorporated by semi-synthesis:
- Impact on the spectrum of activity (beta-lactamases, PBP affinity, etc.)
- Significant role in activity and classification by generation
Adverse Reaction
- Pain at the site of (i.m.) injection.
- Diarrhea and hypersensitivity reactions like penicillins.
- Nephrotoxicity is highest with cephaloridine.
- Platelet dysfunction and bleeding Disulfiram-like reaction.
Uses
- In penicillin-producing staphylococcal infections Example cephalothin.
- Gonorrhea is caused by penicillinase-producing organisms; for Example cefuroxime and cefotaxime.
- Septicemias are caused by gram-negative organisms.
Other β- Lactam Antibiotics
Monobactams: Aztreonam is a monocyclic novel p-lactam antibiotic that has resistance to β-lactamase. It is active against gram-negative bacilli, H. influenza, and Pseudomonas but does not affect gram-positive cocci.
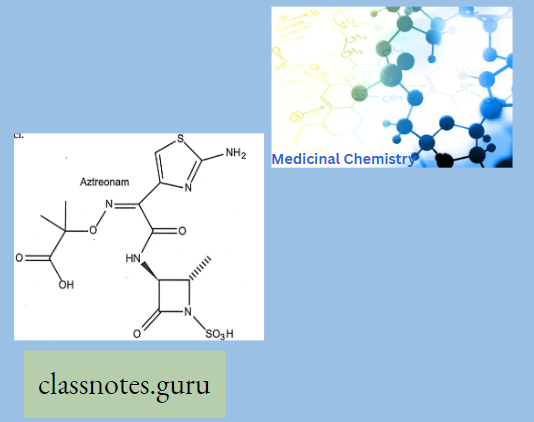
- It is used in patients allergic to penicillin or cephalosporins.
- Adverse effect: hypersensitivity reactions and thrombophlebitis.
Carbapenems
Imipenem:
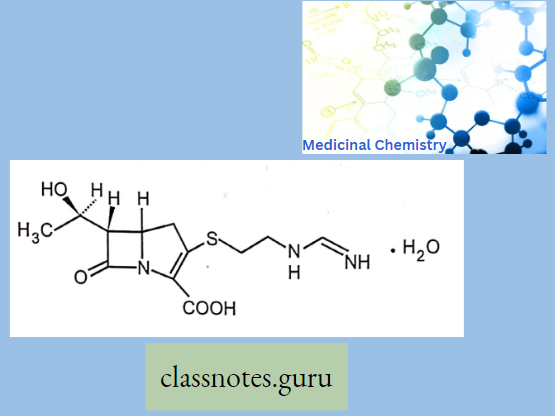
Ertapenem:
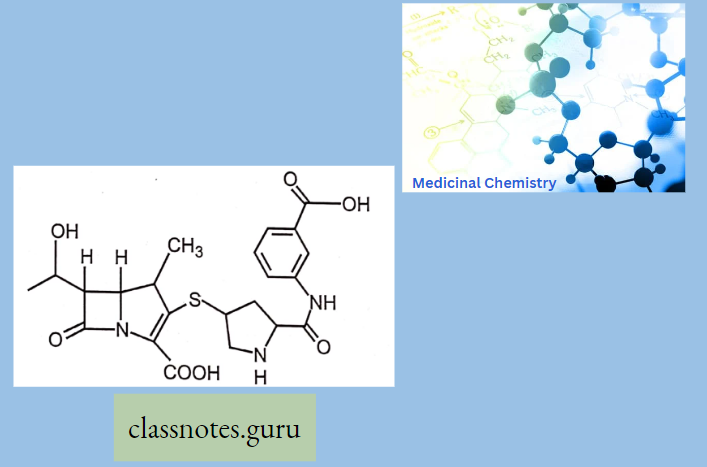
- Penicillin-like, but the sulfur atom of the thiazolidine ring is replaced with a carbon atom.
- These are potent and very broad-spectrum, β-lactam antibiotics. It is resistant to (β- lactamase).
- Unlike Meropenem and Ertapenem, Imipenem is rapidly inactivated by dehydroxypeptidase.
- For this reason, imipenem combined with a dehydroxypeptidase inhibitor called cilastatin, which has similar pharmacokinetics to imipenem
- Probencid inhibits tubular secretion of imipenem-like penicillins.
- Carbapenem exerts cross-sensitivity with penicillins.
- Cephalosporins and other beta-lactams should not be administered to patients who are allergic to these drugs.
- Contraindicated in epileptic patients, higher dosage can produce convulsions.
Antibiotics Multiple Choice Questions
Question 1. Cephalosporin is obtained by
- P. notatum
- P.acrcmonium
- S. Venezuela
- None
Answer:2. P.acrcmonium
Question 2. Cephalosporin is contained.
- Thiazolidine
- Dihydrothiazine
- Purines
- Pyrimidines
Answer:2. Dihydrothiazine
Question 3. The starting material of Cephalosporin…
- 6-amino penicillinic acid
- 6-amino cephalosporin acid
- Phenylalanine
- All
Answer:2. 6-amino cephalosporin acid
Question 4. Cephalosporin excreted by
- Renal tubular secretion
- Sweat secretion
- Both
- None
Answer:1. Renal tubular secretion
Question 5. Which one is first-generation cephalosporin?
- Cefalexin
- Cefoperazone
- Cefepime
- None
Answer: 1. Cefalexin
Question 6. The antimicrobial action of penicillin is the inhibition of
- Protein synthesis
- Cell wall synthesis
- Cell membrane synthesis
- DNA synthesis
Answer:3. Cell membrane synthesis
Question 7. The beta-lactam antibiotics are
- Penicillin
- Cephalosporin
- Imipenem
- All of the above
Answer: 4. All of the above
Question 8. The antimicrobial activity of penicillin is due to
- Thiazolidine ring
- Beta-lactam ring
- 6-APA
- None
Answer: 2. Beta-lactam ring
Question 9. Which of the following is an anti-seudomonal penicillin?
- Carbenicillin
- Methicillin
- Ampicillin
- Azlocillin
Answer: 2. Methicillin
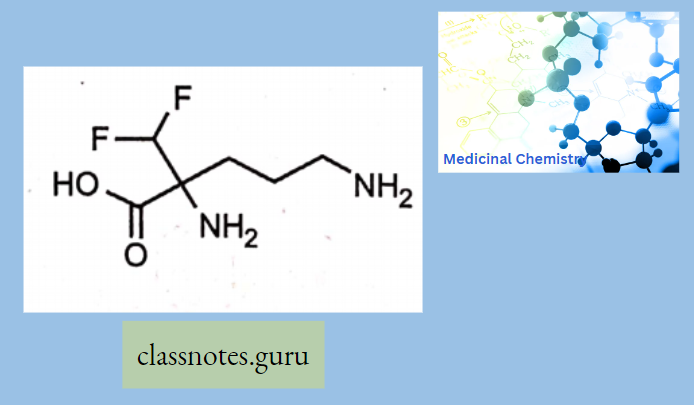
Question 10. How much chiral carbon is present in the penicillin structure?
- 1
- 2
- 3
- 4
Answer:3. 3
Question 11. Which enzyme is responsible for the formation of the cell wall of bacteria?
- Amylase
- Transpetidase
- Mono oxidase
- None
Answer: 2. Transpeptidase
Antibiotics Short Answer Questions
Question 1. Which enzyme is responsible for breaking the beta-lactam ring?
Answer: Beta-lactamase enzyme one is responsible for the breakdown of the penicillin ring.
Question 2. Which ring is present in penicillin?
Answer:
All penicillins are derivatives of 6-aminopenicillins added (thiazolidine ring is attached) to a (β- β-lactam ring that carries a secondary amino group (RNH-)) and contains a beta-lactam ring structure that is essential for antibacterial activity.
Question 3. Write various types of components present in bacterial cell walls.
Answer:
Various Types Of Components Present In Bacterial Cell Walls: The bacterial cell wall is mainly composed of peptidoglycan which is a mucopolysaccharide that mainly contains N- N-acetyl glucosamine and N-acetyl muramic add.
Question 4. Write the mechanism of action of penicillin.
Answer:
The Mechanism Of Action Of Penicillin: They act as an irreversible inhibitor of the enzyme transpeptidase, an enzyme bacteria use to make their cell walls. The final transpeptidation step in the synthesis of the peptidoglycan is facilitated by transpeptidase known as penicillin-binding proteins (PBPs). PBPs bind to the D- Ala-D-Ala at the end of muropeptides, the peptidoglycan precursors to crosslink the peptidoglycan. p-lactam antibiotics mimic the site and competitively inhibit PBP cross-linking of peptidoglycan.
Question 5. Which enzyme is responsible for the cross-linking of NAG and NAM?
Answer:
Enzyme transpeptidase is an enzyme bacteria use to make their cell walls. The final transpeptidation step in the synthesis of the peptidoglycan is facilitated by transpeptidase known as penicillin-binding proteins (PBPs). PBPs bind to the D-Ala-D-Ala at the end of mucopeptides, the peptidoglycan precursors to crosslink the peptidoglycan.
Question 6. Write the mechanism of action of cephalosporin.
Answer:
Mechanism Of Action Of Cephalosporin: They act as an irreversible inhibitor of the enzyme transpeptidase, an enzyme bacteria use to make their cell walls. The final transpeptidation step in the synthesis of the peptidoglycan is facilitated by transpeptidase known as penicillin-binding proteins (PBPs).
Question 7. Write the specific feature of aztreonam.
Answer:
The Specific Feature Of Aztreonam: It is a monocyclic novel (β-lactam antibiotic that has resistance to (β-lactamase). It is active against gram-negative bacilli, H. influenza, and Pseudomonas but does not affect gram-positive cocci.
Question 8. Write the example of a third-generation cephalosporin.
Answer:
An Example Of A Third-Generation Cephalosporin: Ceftriaxone, Cefoperazone, Cefotaxime
Question 9 . Write the example of carbapenems.
Answer:
Example Of Carbapenems: Imipenem, Meropenem, Ertapenem
Question 10. Write the uses of Cephalosporins.
Answer:
Uses Of Cephalosporins
- In penicillin-producing staphylococcal infections Example, cephalothin.
- Gonorrhea is caused by penicillinase-producing organisms; for Example cefuroxime and cefotaxime.
- Septicemias are caused by gram-negative organisms.