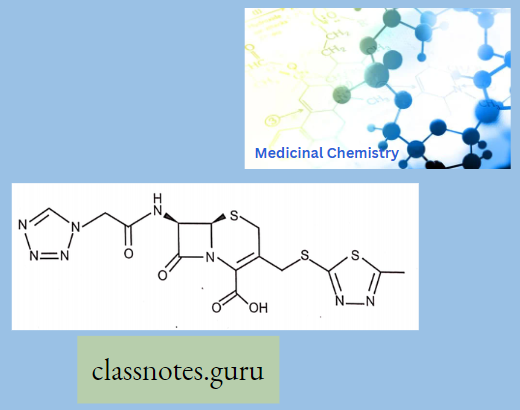
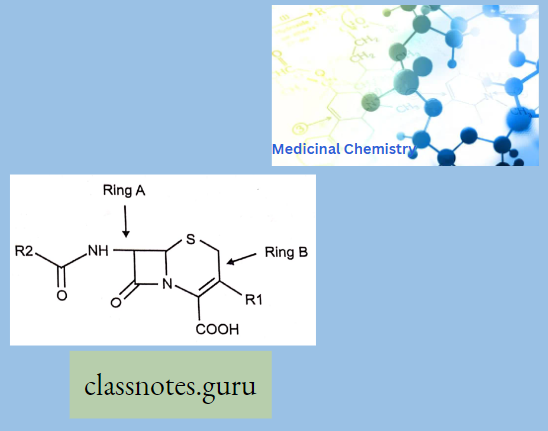
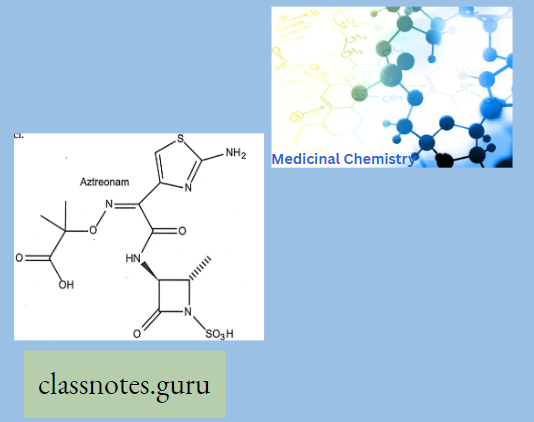
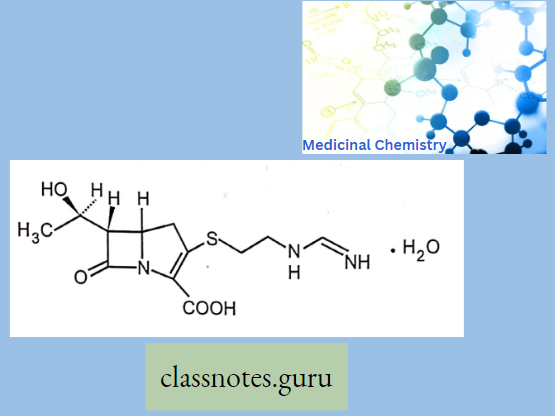
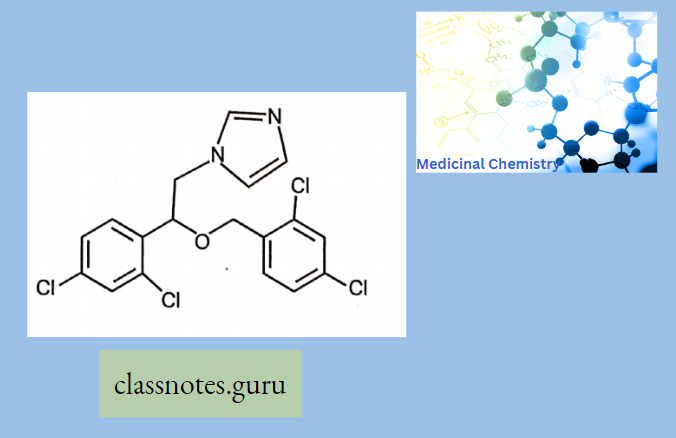
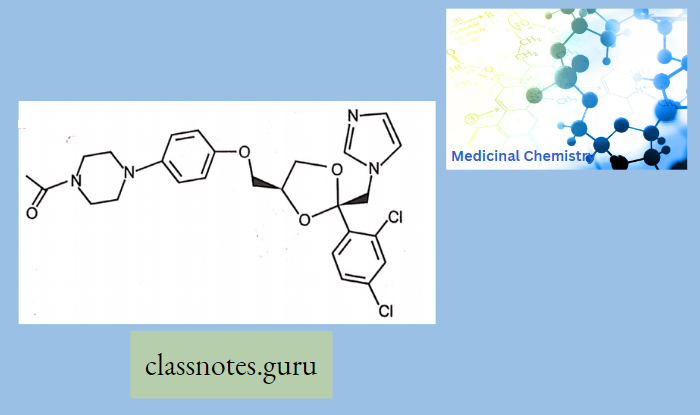
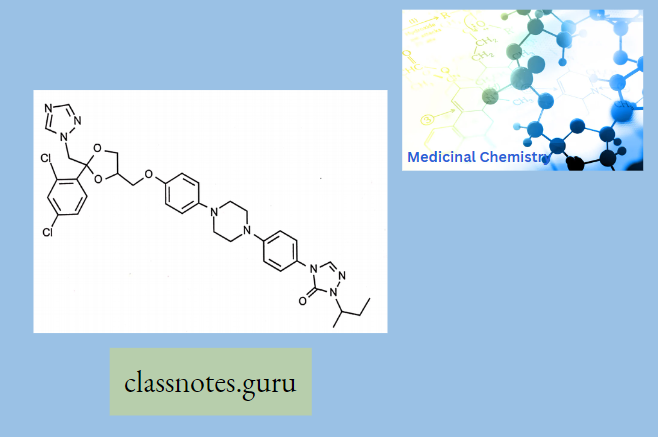
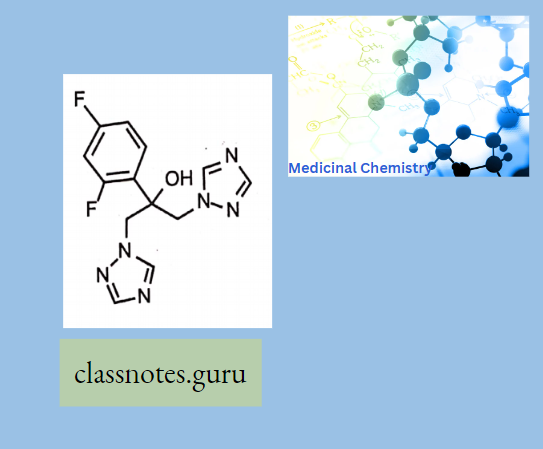
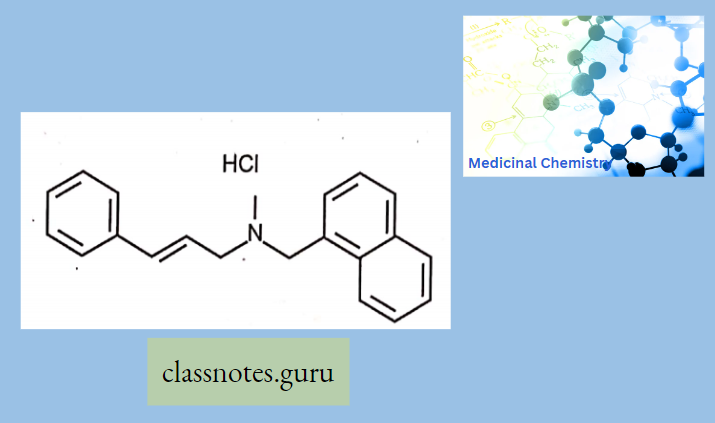
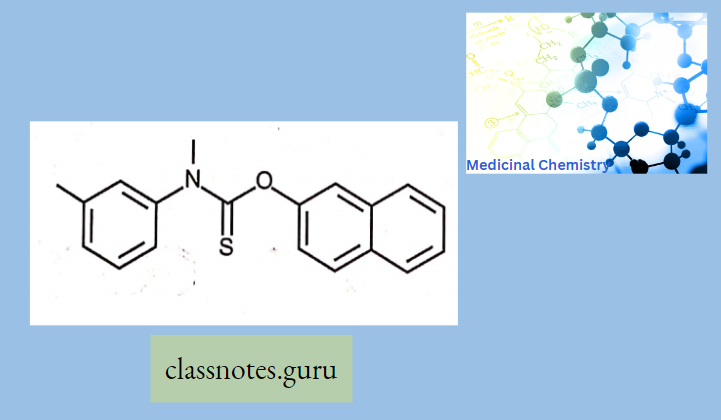
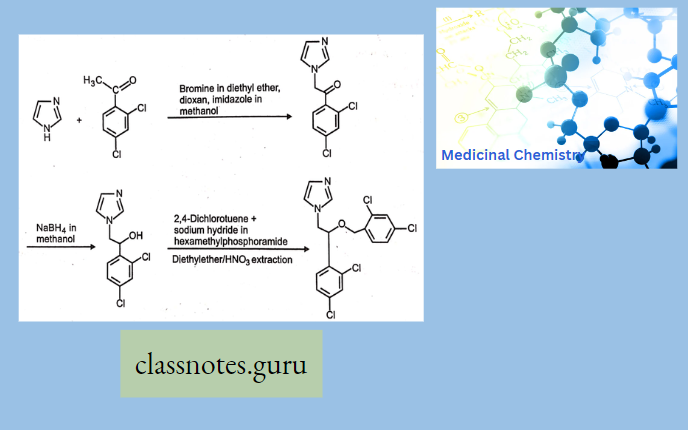
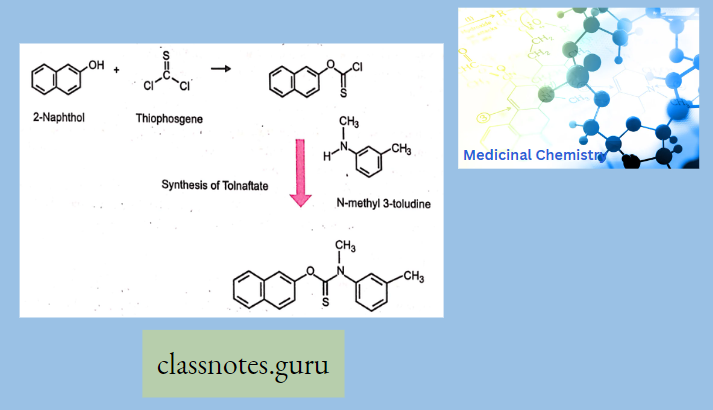
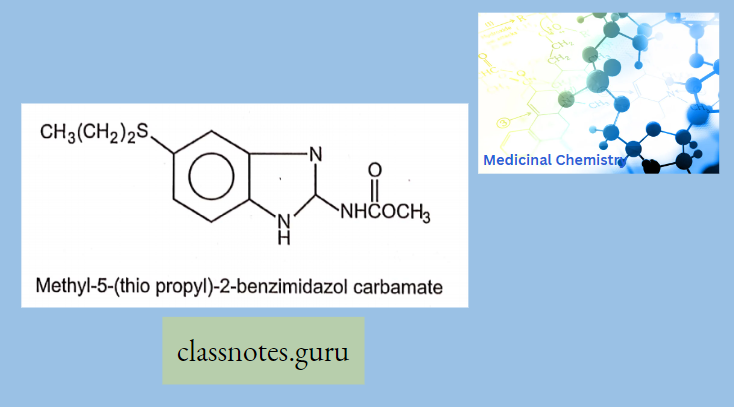
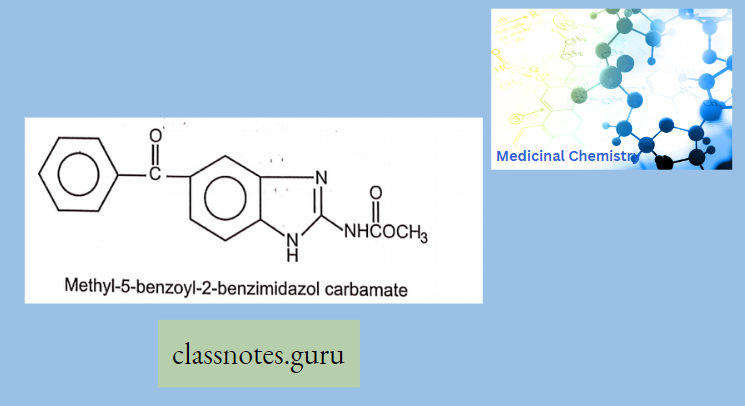
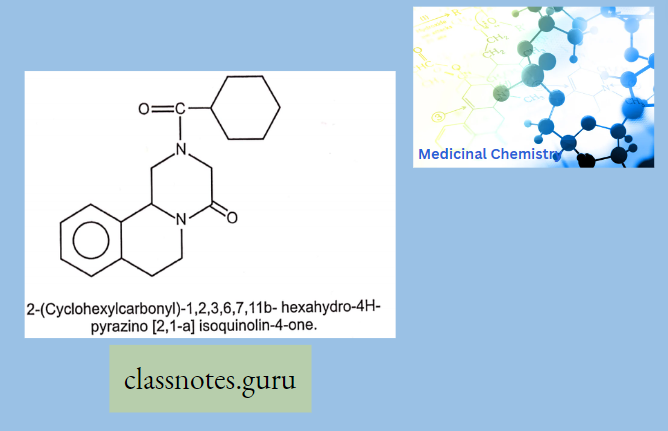
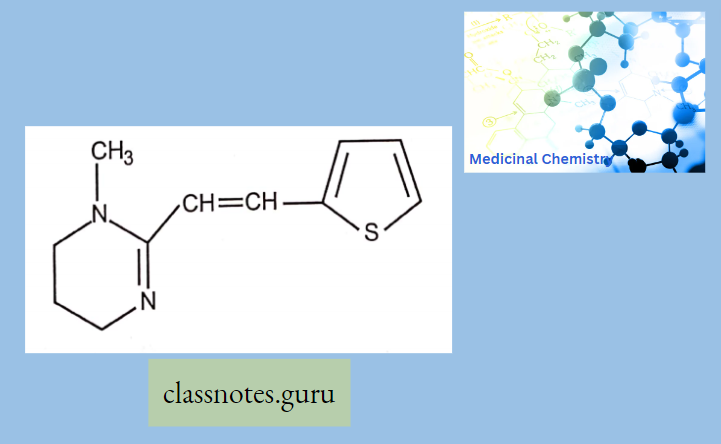
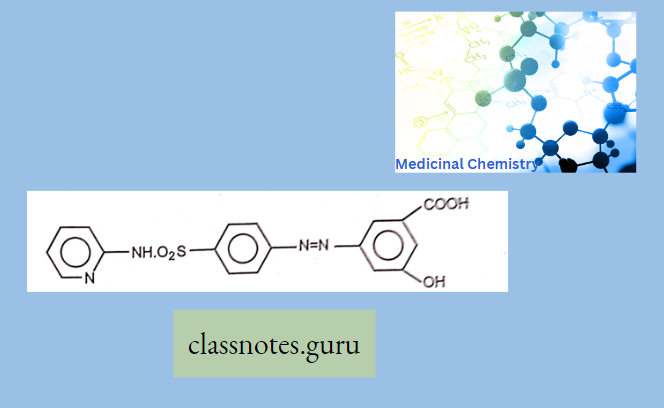
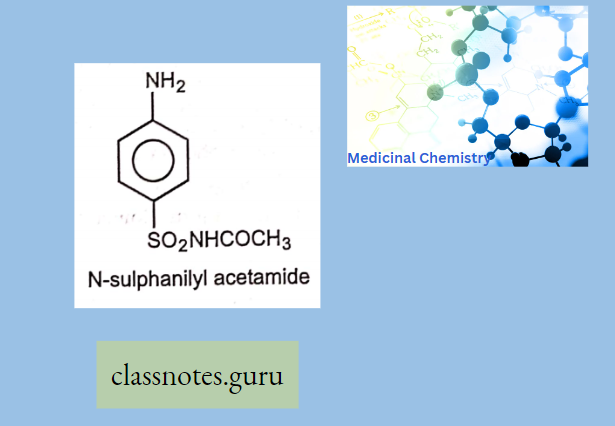
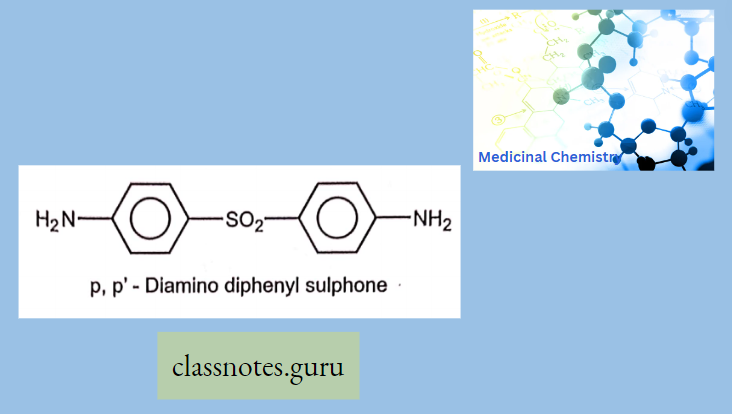
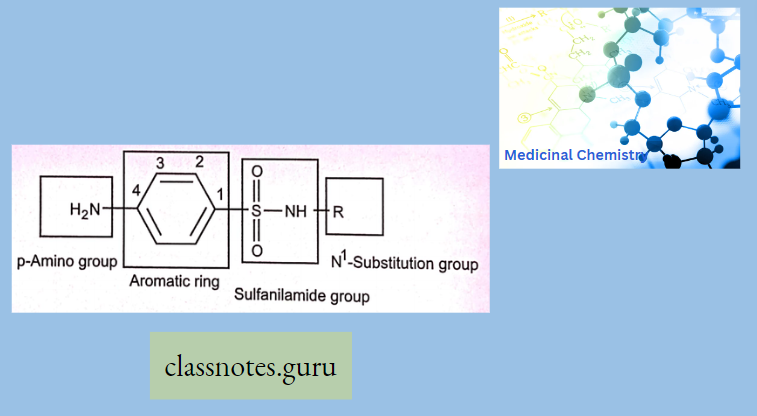
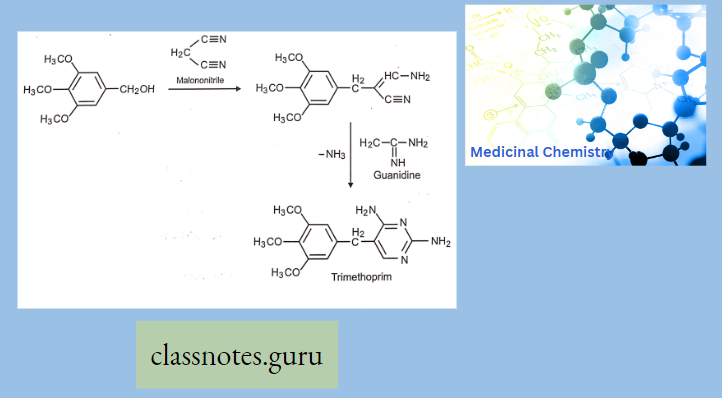
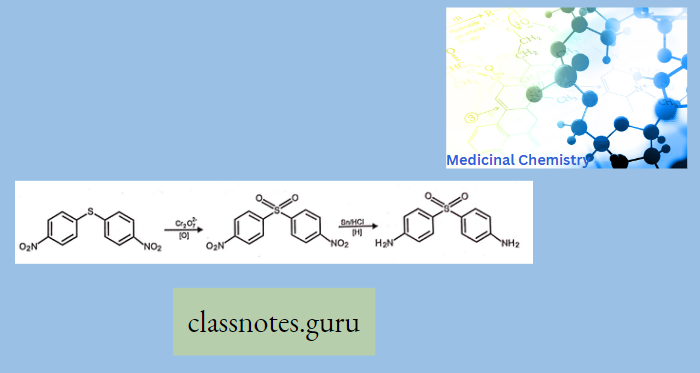
Macrolide Antibiotics Introduction
Macrolides belong to one of the most commonly used families of clinically important antibiotics used to treat infections caused by Gram-positive bacteria such as Staphylococcus aureus, Streptococcus pneumonia, and Streptococcus pyogenes.
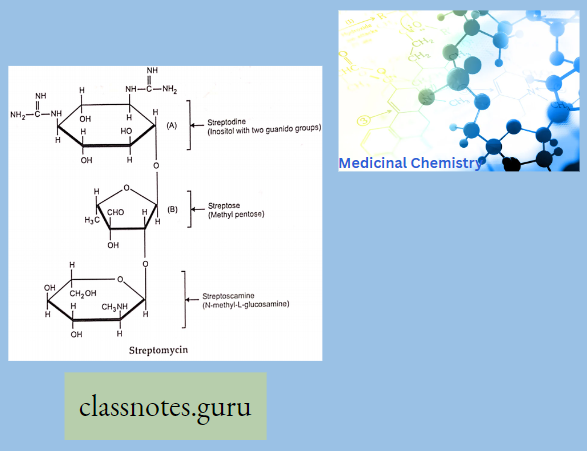
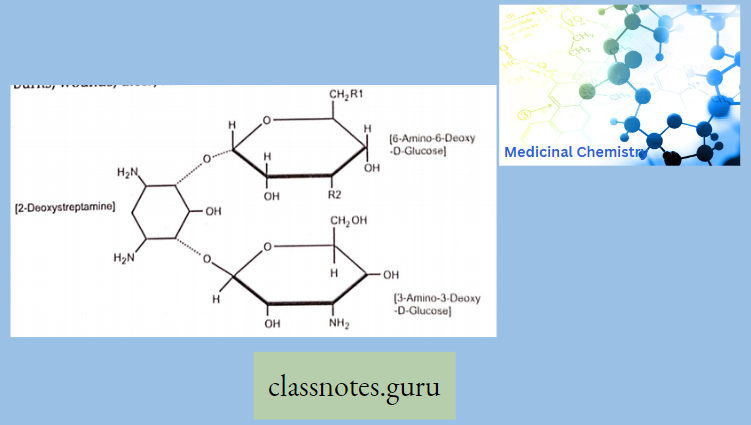
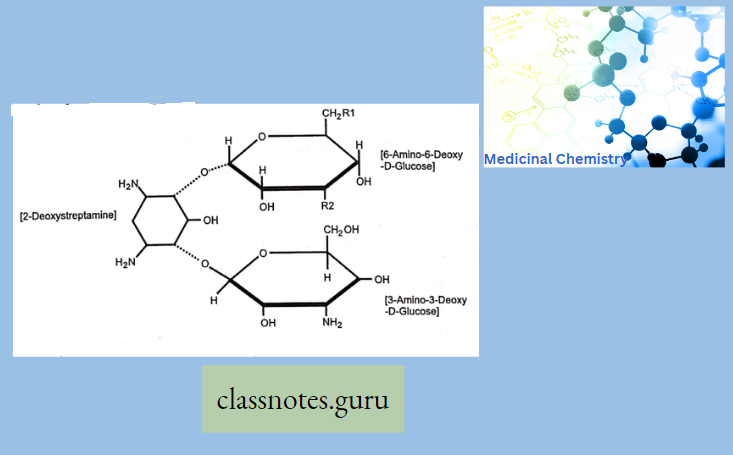
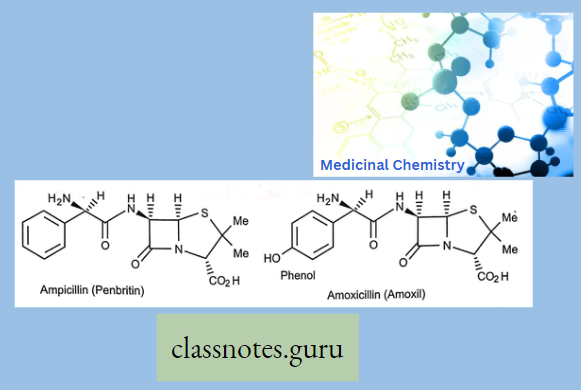
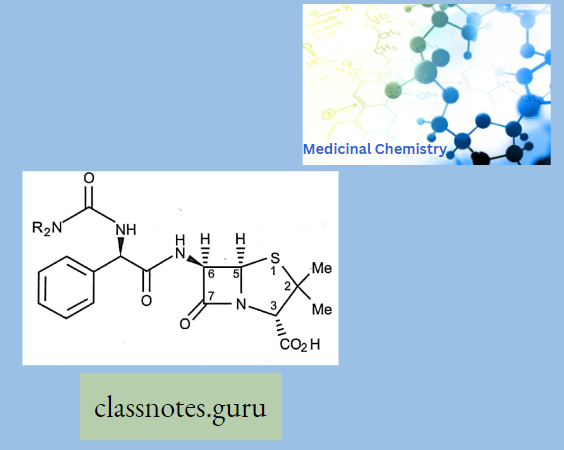
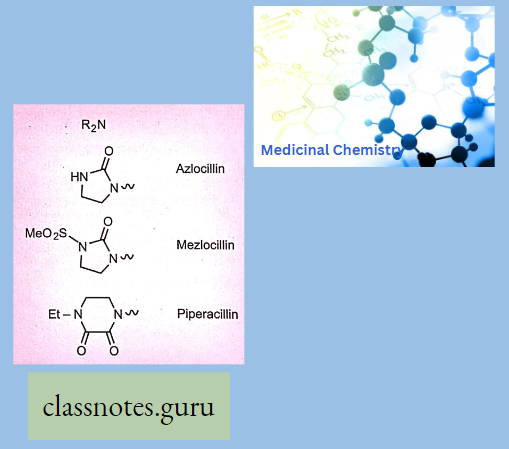
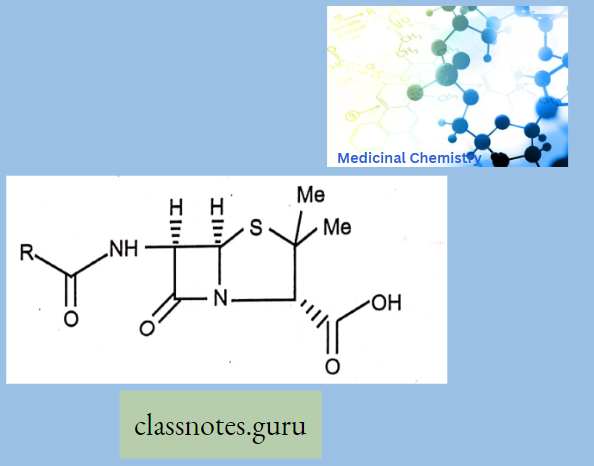
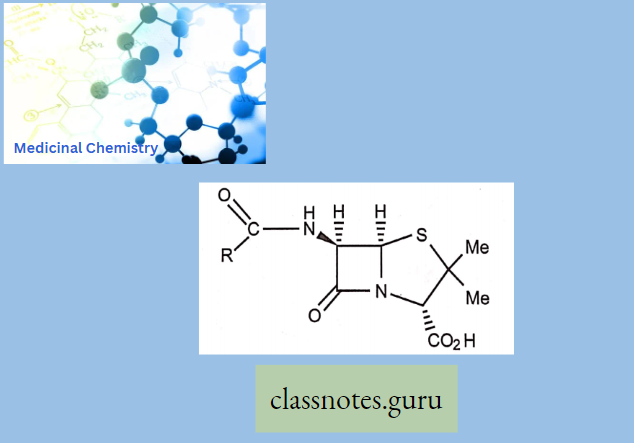
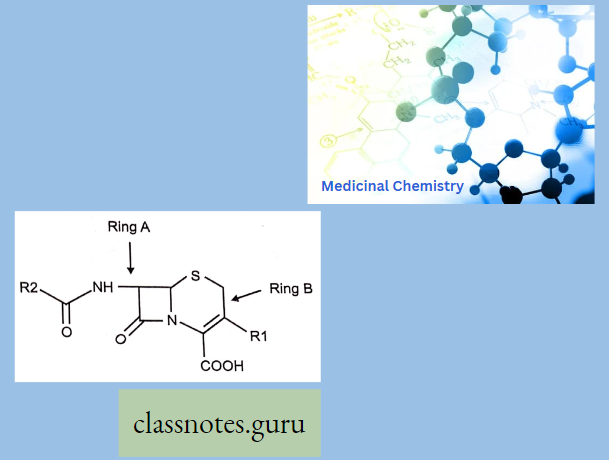
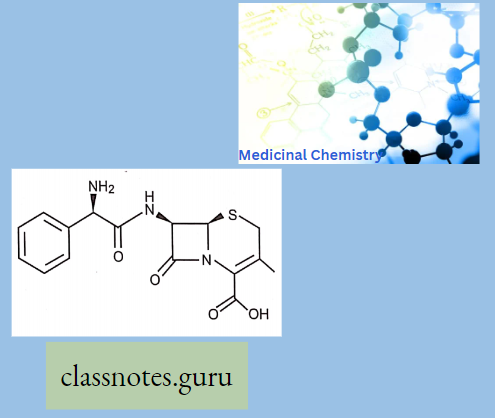
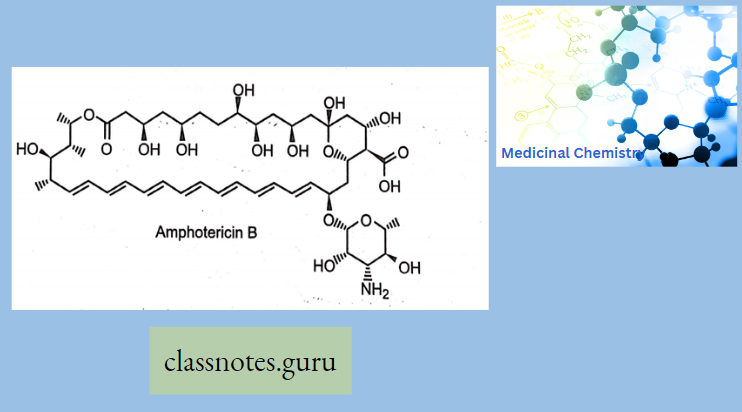
Chemically, macrolides are represented by a 14-, 15- or 16-membered lactone ring carrying one or more sugar moieties and additional substitutions linked to various atoms of the lactone ring
Macrolide Antibiotics Mechanisam Of Action
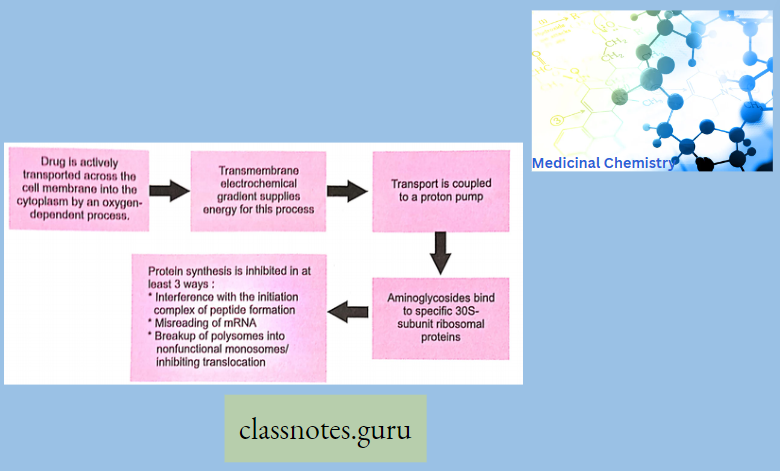
Bind to 50s subunit of ribosome causing them to dissociate from the mRNA resulting in premature termination of the amino acid chain and cessation of protein synthesis.
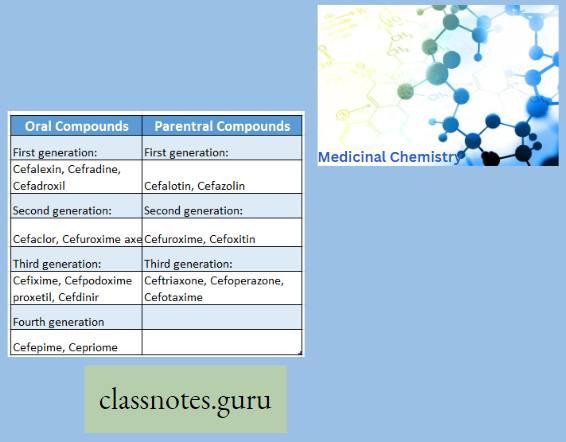
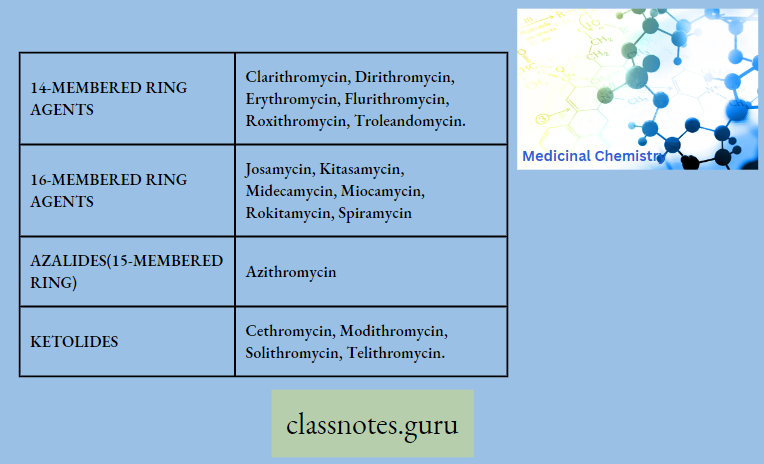
Macrolide Antibiotics Classification
Pharmacokinetics
Tacrolidc antibiotics are fairly absorbed from the gastrointestinal tract They penetrate moot tissues and host cells excellently. The concentrations in phagocytic cells exceed peach maximum serum levels by several folds.
- On me other hand, macrolides penetrate poorly into the brain, synovial fluid, and fetal tissues, macrolide antibiotics are excreted into mucosal fluid, breast milk, bile, and urine. The ratio of urinary or fecal excretion is variable.
- The portion of macrolide antibiotics excreted into bile is partially reabsorbed in the gut enterohepatic circulation,) Some drug is metabolized in the liver as well.
Read and Learn More Medicinal Chemistry III Notes
Spectrum Of Activity
- Active against give bacilli and give cocci
- Also active against H-Influenza, mycoplasma pneumonia, N.Gonorrhoea, and legionella
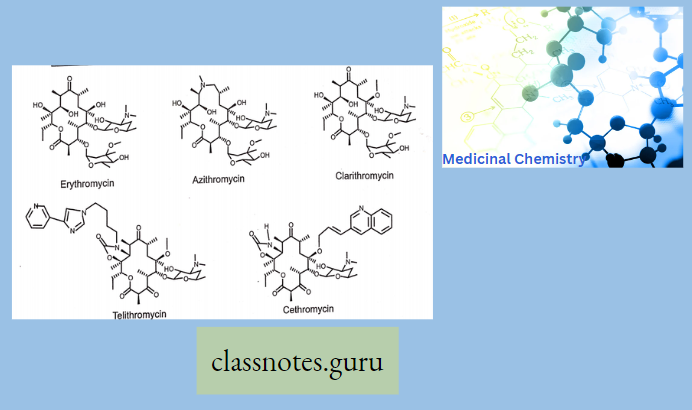
Chemistry Of Macrolides
The commercial product is Erythromycin A which is different from Erythromycin B in having -OH group at 12 position of aglycon.
- Erythronolide-Aglycon part of Erythromycin
- Glycon part-1. Basic ring-Desosamine 2. Neutral ring-cladinose While in the case of Erythromycin C, it has Mycarose as a neutral glycogen part instead of Cladinose
- It acts as an Enzyme inhibitor (Cyto-P-450 oxidase) for other drugs.
- Like Theophylline, Hydroxy coumarine, Benzodiazepine (Alprazolam, Midazolam), carbamazepine. Cyclosporine is an antihistaminic drug While the activity of terfenadine and astemizole is potentiated by Erythromycin.
- The stability of Erythromycin is at or neutral pH (7)
- Clarithromycin-6-methyl ether derivative of Erythromycin. (6-OH group is methylated to 6-OCH3).
- It acts as an Enzyme inhibitor (Cyto-P-4aO oxidase) for other drugs.
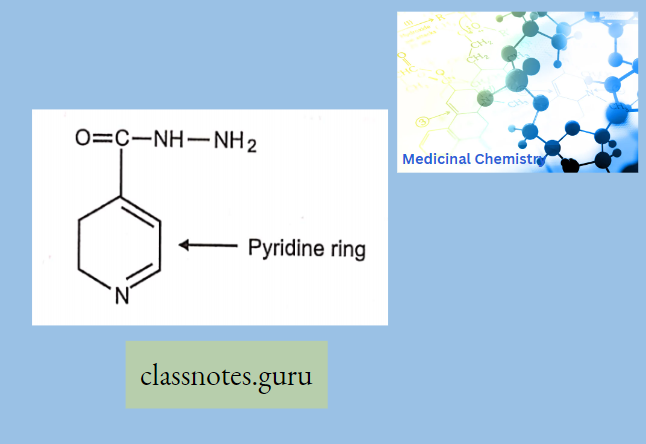
- Specifically used to treat Lyme disease caused by Borrelia Burdorferi.
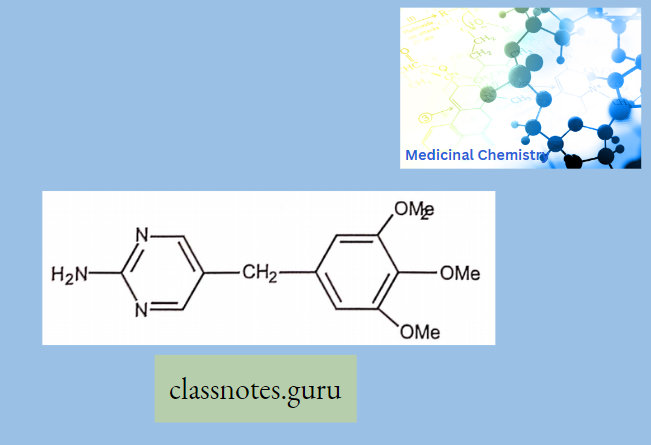
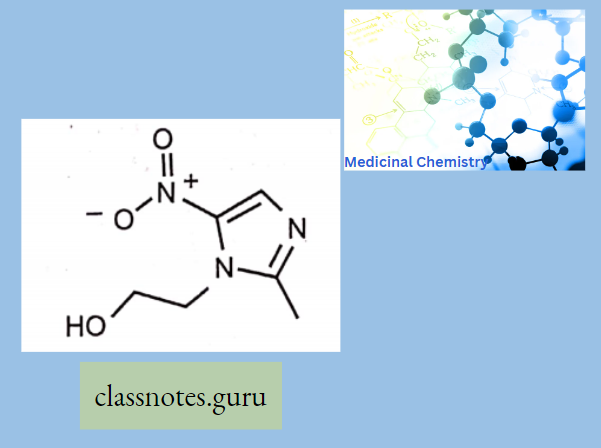
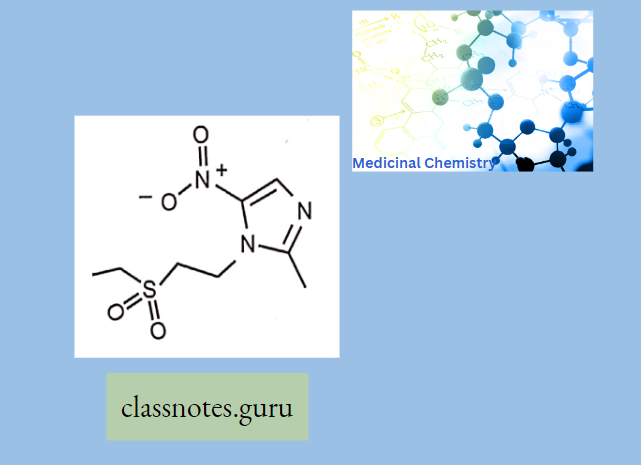
- Azithromycin, prepared by Beckmann rearrangement of 9-Oxime followed by N-methylation and reduction of resulting ring-expanded lactam. Nitrogen-containing 15-membered rings Macrolide is known as Azalides.
- It does not act as an enzyme inhibitor (Cyto-P-450 oxidase) for other drugs.
- Removal of 9-keto group-increasing stability of azithromycin to acid-catalyzed degradation. These changes also increase lipid solubility.
- Dirithromycin-Having 9N, 11 O-Oxazine ring
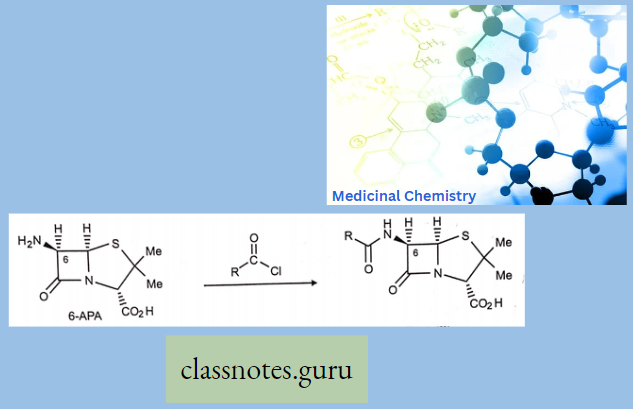
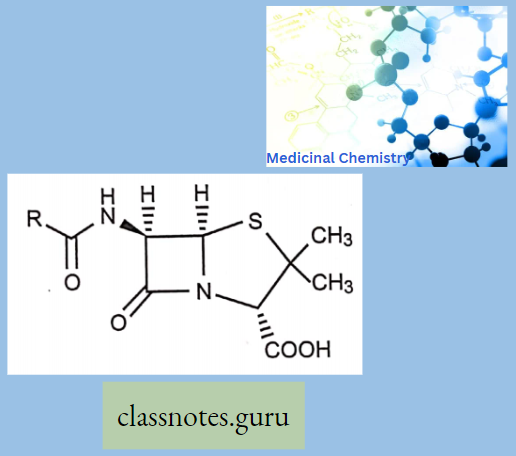
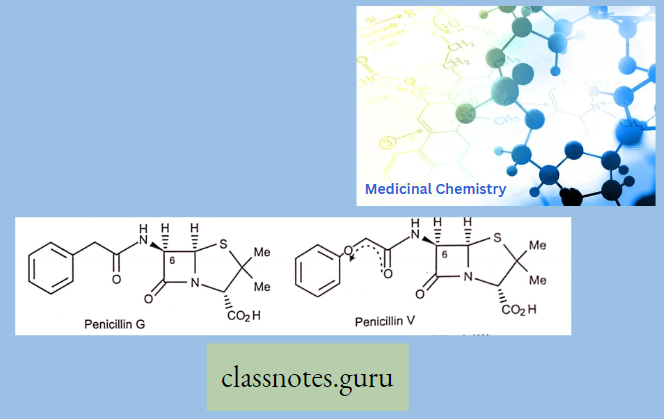
Sar
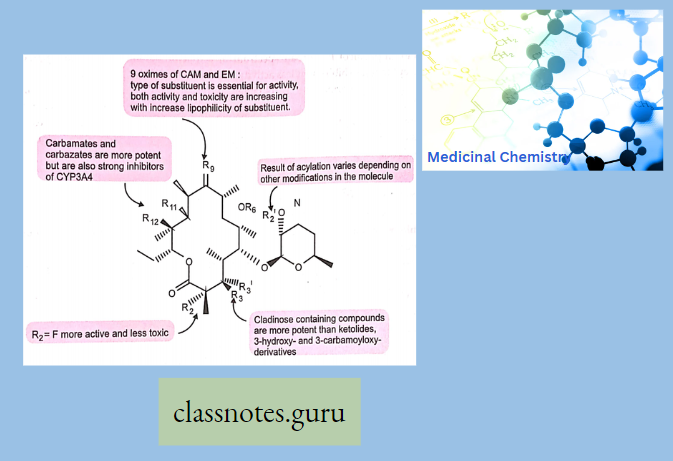
Chloramphenicol
Chloramphenicol is a broad-spectrum antibiotic with bacteriostatic activity and a wide spectrum of activity but is currently a backup drug for infections due to Salmonella typhi, B. fragilis, Rickettsia, and possibly bacterial meningitis.
- Chloramphenicol was initially obtained from Streptomyces Venezuela
- Chloramphenicol has a broad spectrum of activity resembling that of the tetracyclines except that it exhibits a bit less activity against some gram-positive bacteria.
- Chloramphenicol contains chlorine and is obtained from an actinomycete, and thus, named Chloromycetin.
- Chloramphenicol is specifically recommended for the treatment of serious infections caused by H. influenza, S. typhi (typhoid), S. pneumoniae, and N. meningitides.
- Its ability to penetrate the CNS presents an alternative therapy for meningitis and exhibits anti-rickettsial activity.
Chloramphenicol The Adverse Effects: Chloramphenicol causes bone marrow depression and fatal blood dyscrasias.
Chloramphenicol Properties:
- Chloramphenicol is a white or greyish-white or yellowish-white crystalline powder or fine crystals, slightly soluble in water, soluble in alcohol, and propylene glycol.
- Chloramphenicol was the first and still is the only therapeutically important antibiotic to be produced in competition with microbiological processes.
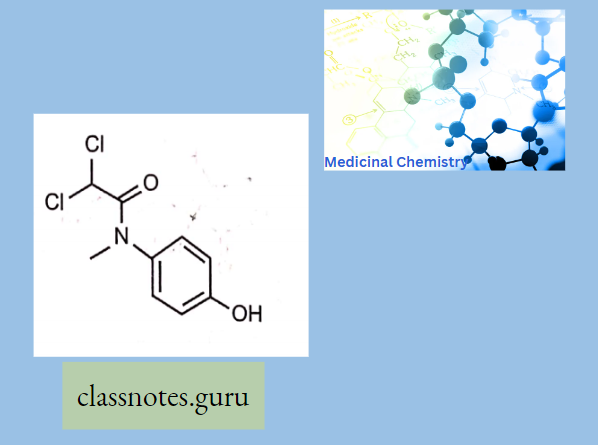
- Chloramphenicol contains a nitrobenzene moiety and is a derivative of dichloroacetic acid. Since it has two chiral centers, four isomers are possible.
- The D is the biologically active form.
Chloramphenicol Mechanism Of Action
- Chloramphenicol is a bacteriostatic by inhibiting protein synthesis.
- Chloramphenicol prevents protein chain elongation by inhibiting the peptidyl transferase activity of the bacterial ribosome.
- Chloramphenicol specifically binds to A2451 and A2452 residues in the 23S rRNA of the 50S ribosomal subunit, preventing peptide bond formation.
- Chloramphenicol palmitate Prodrug is designed to mask the bitter taste
- Chloramphenicol succinate Prodrug designed to increase water solubility
Chloramphenicol Adverse Effects
- Dose-dependent bone marrow suppression is common
- Aplastic anemia is rare (1 in 35, 000).
- Gray baby syndrome in neonates (decreases glucuronysyl transferase)
- Optic neuritis in children.
- Broad spectrum antibiotic
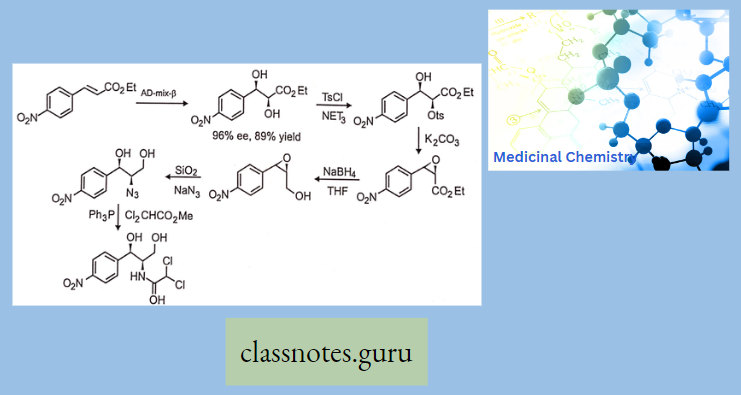
- Nowadays, it is prepared by synthetic route from p-Nitro acetophenone.
Chemistry Of Chloramphenicol:
- Chloramphenicol has two chiral carbons, so a total of four (4) isomers are possible D-erythro, L-L-erythro, D-threo, and L-threo.
- Among these four isomers, the D-three isomer is the most active. The prodrug of Chloramphenicol viz., Chloramphenicol palmitate (USP) which is a tasteless product is intended for pediatric usage.
Sar Of Chloramphenicol
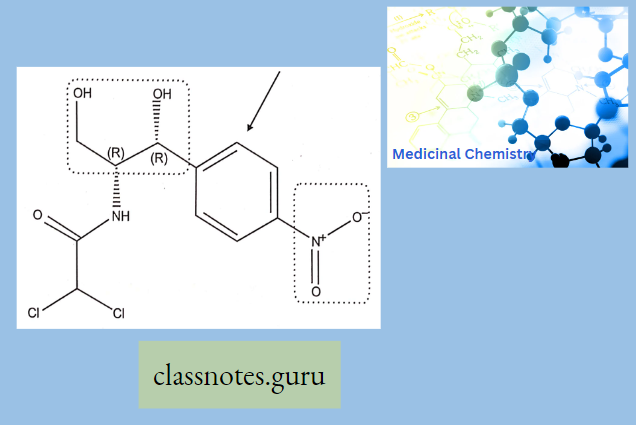
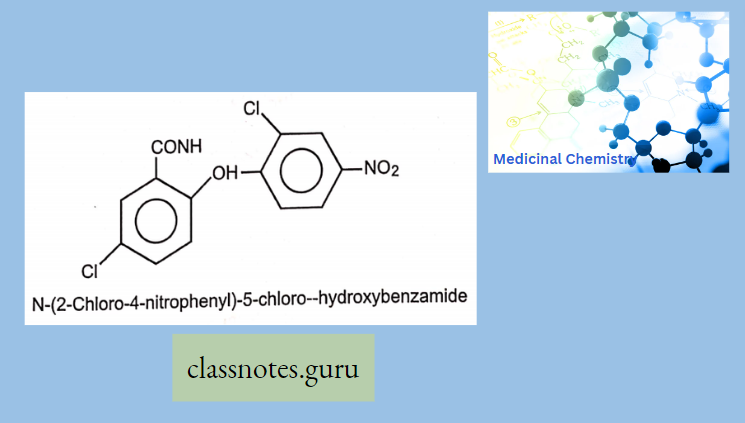
Modification Of The p-Nitrophenyl Group:
The para-nitrophenyl group may be modified through the following ways:
- Replacement of the nitro group by other substituents leads to a reduction in activity.
- Shifting of the nitro group from the para position also reduces the antibacterial activity.
- Replacement of the phenyl group by the alicyclic moieties results in less potent compounds.
Modification Of Dichloro Acetamido Side Chain: Other halo derivatives of the side chain are less potent although major activities are retained.
Modification Of 1,3-propanediol: If the primary alcoholic group on the C-l atom is modified, it results in a decrease in activity; hence, the alcoholic group seems to be essential for activity.
Metabolism
Major Route: Formation of 3-O-Glucrodination
Minor Route: Reduction of the p-Nitro group to amino
Metabolism Use
- Meningitis
- Active against gm+ve and gm-ve bacteria that are resistant to PenicillinG and ampicillin.
- Active against H.Influenza, S.Typhi, S.Pneumonia, B.fragilis and N.meningitis
- In UTI
- Rickettsial infections as “Rocky Mountain Spotted Fever”
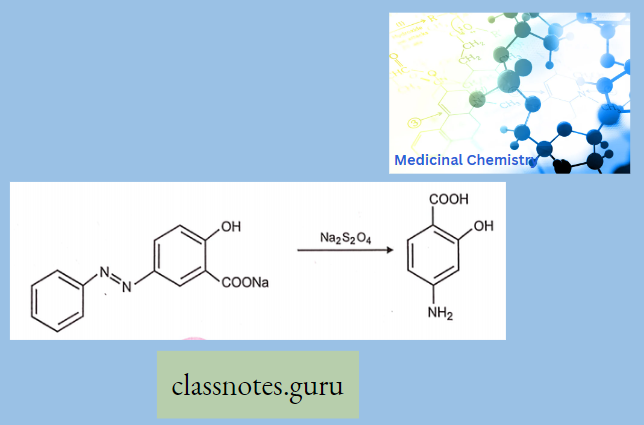
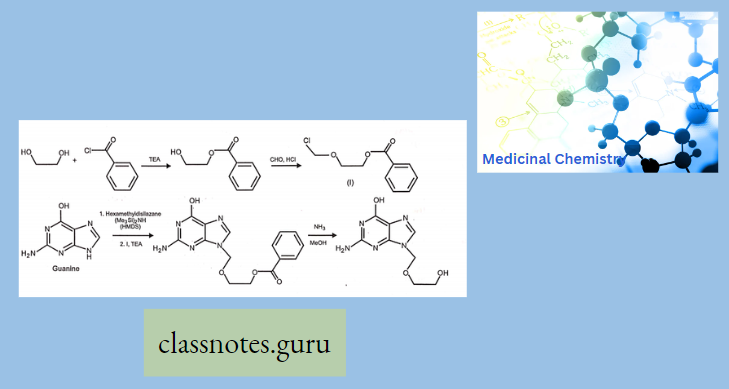
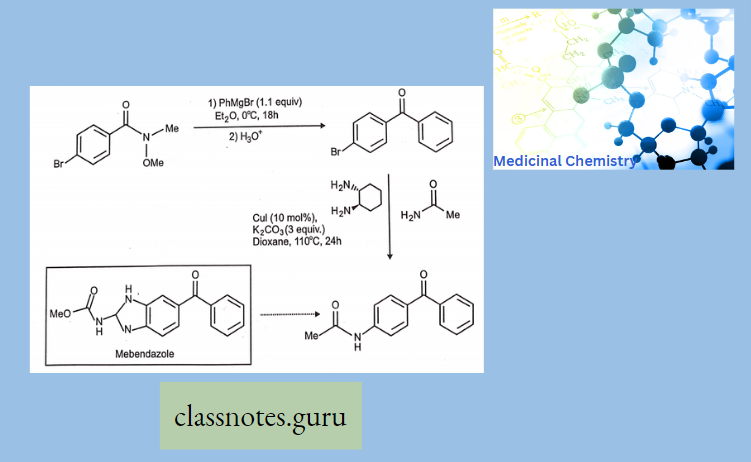
Synthesis Of Chloramphenicol
Vancomycin
Vancomycin is an antibiotic that has been around for the last forty years. Recently, however, vancomycin has been increasing in popularity because of its effectiveness in treating resistant organisms. One of two glycopeptide antibiotics in clinical use, vancomycin has a unique chemical structure.
Vancomycin is comprised of a glycosylated hexapeptide chain containing unusual amino acids. In addition, vancomycin is fairly rigid because of aromatic rings that are halogenated and crosslinked by aryl ether bonds.
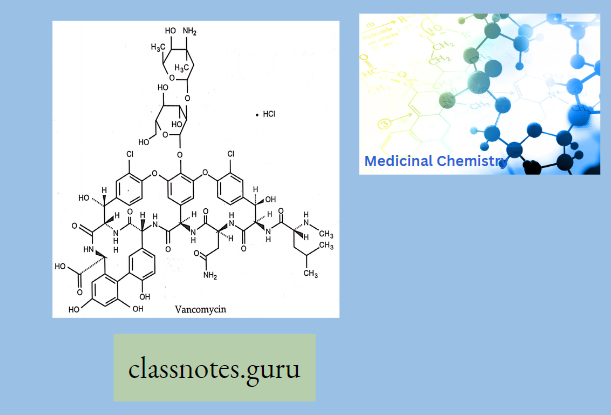
Vancomycin can penetrate the gram-positive cell wall and therefore is effective against gram-positive organisms. Vancomycin shows no activity against gram-negative organisms, however, because it is unable to penetrate the gram-negative cell wall.
Vancomycin Mechanism Of Action
- Vancomycin acts by inhibiting bacterial cell wall biosynthesis. Specifically, vancomycin binds to the D-Alanyl-D-Alanine portion of the dipeptide, a key component for the transpeptidase reaction, and forms three hydrogen bonds.
- By covering the substrate for cell wall transaminase, vancomycin prevents the molecule from being transported to the cell wall. Cross-linking does not occur and the integrity of the bacterial cell wall is compromised. The bacterial cell cannot withstand changes in osmotic pressure and it will rupture and die. Vancomycin is bactericidal.
Use Of Vancomycin: Vancomycin is FDA-approved for the treatment of several bacterial infections, including infections caused by susceptible staphylococcus, streptococcus, enterococcus, and diphtheroid organisms. Vancomycin is commonly used in clinical practice to treat endocarditis and meningitis.
Vancomycin Adverse Effects: The most common side effects associated with vancomycin include nausea and vomiting. Rarely, nephrotoxicity, ototoxicity, and neutropenia.
Vancomycin Drug Interactions: Many drugs may increase the adverse effects of vancomycin. Medicines that affect the kidneys, such as aminoglycosides, may increase the risk of kidney damage and should be avoided.
- In addition, co-administration of vancomycin and succinylcholine may result in a prolonged neuromuscular blockade.
- Patients should be monitored and a dose adjustment may be necessary.
- Vancomycin also has been reported to moderately interact with warfarin, potentially increasing the risk of bleeding.
- Patients who take warfarin should be closely monitored as they initiate and stop vancomycin therapy.
Clindamycin
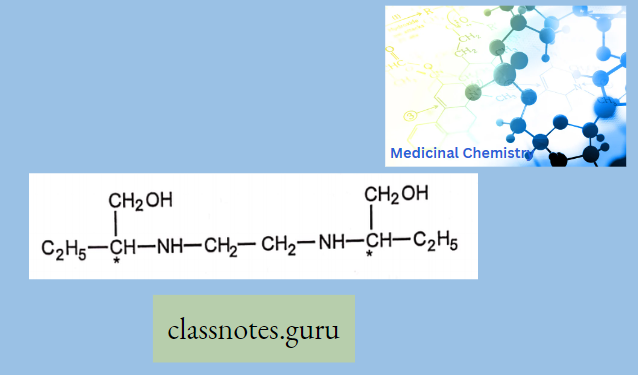
Clindamycin belongs to a class called Lincomycins which were isolated from Streptomyces. This class resembles another antibiotic class called Macrolides and is active against both gram-negative, and gram-positive bacteria, and anaerobes.
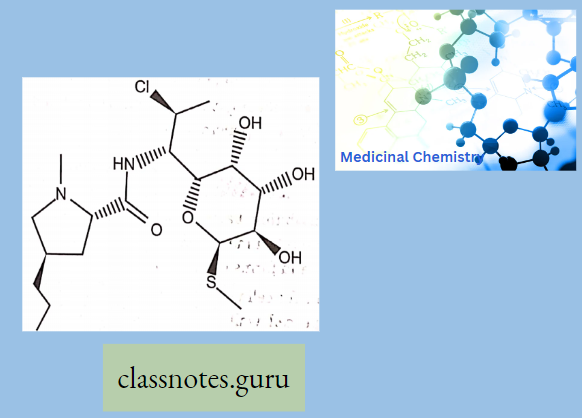
Lincomycin
Lmcomycin shows cross-resistance with other Macrolides and Stre’ptoGramins because they bmd to the ribosome m the same way. Lincomycins are water soluble and when given as a HC1 salt they can distribute to other body tissues.
Lincomycin Mechanism Of Action: The Lincomycins, like the Macrolides, are bacteriostatic. They act by binding to the 50S ribosomal subunit and causing the release of a fragmentary peptide by preventing the translocation of peptidyl-tRNA from the A-site to the P-site. By inducing the formation of these incomplete peptides, the growth of the bacterial cell is inhibited.
Use Of Clindamycin
Clindamycin is FDA-approved for the treatment of bacterial infections due to Staphylococcus aureus, Staphylococcus epidermidis, and Streptococcus pyogenes, as well as for the treatment of acne vulgaris, bacterial vaginosis, and pelvic inflammatory disease.
Clindamycin Adverse Effects
- The most common adverse effects reported with clindamycin use are diarrhea, nausea, and rash. In rare cases, clindamycin may cause pseudomembranous enterocolitis.
- Clindamycin may also affect the liver, resulting in jaundice and increased liver function tests. Clindamycin is contraindicated in patients with an allergy to clindamycin or lincomycin.
Macrolide Antibiotics Multiple Choice Question And Answers
Question 1. The commercial product of macrolides is…
- Erythromycin A
- Erythromycin B
- Erythromycin C Macrolides act on…
- Erythromycin D
Answer: 1. Erythromycin A
Question 2. Macrolide acts on.
- Erythromycin A
- 30s ribosome
- Both
- None
Answer: 1. Erythromycin A
Question 3. Which macrolides contain a 15-membered ring in their structure …
- Erythromycin
- Azithromycin
- Clarithromycin
- Roxithromycin
Answer: 2. azithromycin
Question 4. Dopamine is a.
- Acidic ring
- Basic ring
- Neutral
- None
Answer: 2. Basic ring
Question 5. Phototoxicity is the adverse effect of
- Chlortetracycline
- rolitetracycline
- Demeclocycline macrolide acts on
- doxycycline
Answer: 3. Demeclocycline macrolide acts on
Question 6.Macrolide act on
- 50s subunit
- 30s subunit
- 80s subunit
- 70s Subunit
Answer: 1. 50s subunit
Question 7. Chloramphenicol act on
- 50s subunit
- 30s subunit
- 80s subunit
- 70s subunit
Answer: 2.30s subunit
Question 8. Adverse effect of chloramphenicol is
- Gray baby syndrome
- Fanconi syndrome
- Ototoxicity
- Red man syndrome
Answer: 1. Gray baby syndrome
Question 9. The drug of choice for Rocky Mountain spotted
- Chloramphenicol
- Aminoglycosides
- tetracycline
- Macrolide
Answer: 1. Chloramphenicol
Question 10. Vancomycin acts on
- D-Alanyl-D-Alanine
- L- Alanyi D- Alanyl-D-Alanineboth
- Both
- None
Answer: 1. D-Alanyl-D-Alanine
Macrolide Antibiotics Short Question And Answers
Question 1. Classify 14 membered macrolides
Answer:
14 Membered Macrolides
- Clarithromycin
- Dirithromycin
- Flurithromycin
- Troleandomyci
- Erythromycin
- Roxithromycin
Question .2 Write In mime ketolides.
Answer:
In Mime Ketolides
- Modithromycin
- Telithromycin
- Cethromycin
- Modithromycin
- Telithromycin
- Solithromycin
Question 3. What In the mode of action of macrolides?
Answer:
The Mode Of Action Of Macrolides: Bind 509 Hiibunil of ribosome causing them to dissociate from the mRNA resulting in premature termination of the amino acid chain and cessation of protein synthesis.
Question 4 . How does azithromycin degrade?
Answer:
Removal of 9-keto group-increasing stability of azithromycin to acid-catalyzed degradation. These changes also increase lipid solubility.
Question 5. What is the activity spectrum of macrolides?
Answer:
The Activity Spectrum Of Macrolides: Active against gm+ve cocci, bacilli, and gm-ve cocci Also active against H.Influenza, mycoplasma pneumonia, N.Gonorrhoea, and legionella
Question 6. Write the mode of action of macrolide.
Answer:
Mode Of Action Of Macrolide: Bind to 50s subunit of ribosomes causing them to dissociate from the mRNA resulting in premature termination of the amino acid chain & cessation of protein synthesis.
Question 7. Write the mode of action of chloramphenicol.
Answer:
Mode Of Action Of Chloramphenicol: Chloramphenicol is a bacteriostatic by inhibits protein synthesis. It prevents protein chain elongation by inhibiting the peptidyl transferase activity of the bacterial ribosome. It specifically binds to A2451 and A2452 residues in the 23S rRNA of the 50S ribosomal subunit, ‘preventing peptide bond formation.
Question 8. Write the adverse effect of chloramphenicol.
Answer:
The Adverse Effect Of Chloramphenicol: Aplastic anemia is rare Gray baby syndrome in neonates (decreases glucuronysyl transferase) optic neuritis in children.
Question 9. Write the adverse effects of clindamycin.
Answer:
The Adverse Effects Of Clindamycin: The most common adverse effects reported with clindamycin use are diarrhea, nausea, and rash. In rare cases, clindamycin may cause pseudomembranous enterocolitis.
Question 10 Write the mode of action of clindamycin.
Answer:
The Mode Of Action Of Clindamycin: They act by binding to the 50S ribosomal subunit and causing the release of a fragmentary peptide by preventing the translocation of peptidyl-tRNA from the A-site to the P-site. By inducing the formation of these incomplete peptides, the growth of the bacterial cell is inhibited.
Question 11. Write the mode of action of vancomycin.
Answer:
The Mode Of Action Of Vancomycin: Vancomycin acts by inhibiting bacterial cell wall biosynthesis. Specifically, vancomycin binds to the D-Alanyl-D-Alanine portion of the dipeptide, a key component for the transpeptidase reaction, and forms three hydrogen bonds. By covering the substrate for cell wall transaminase, vancomycin prevents the molecule from being; transported to the cell wall.